
Personalised proteome analysis by means of protein microarrays made from individual patient samples
DNA sequencing has advanced to a state that permits studying the genomes of individual patients as nearly a matter of routine. Towards analysing at a tissue's protein content in a similar manner, we established a method for the production of microarrays that represent full-length proteins as they are encoded in individual specimens, exhibiting the particular variations, such as mutations or splice variations, present in these samples. From total RNA isolates, each transcript is copied to a specific location on the array by an on-chip polymerase elongation reaction, followed by in situ cell-free transcription and translation. These microarrays permit parallel analyses of variations in protein structure and interaction that are specific to particular samples.
Apart from procedural information, details are provided on the overall quality of protein microarrays (e.g. percentage of full-length molecules and the structural integrity of proteins) and their perfomance in various forms of analysis.
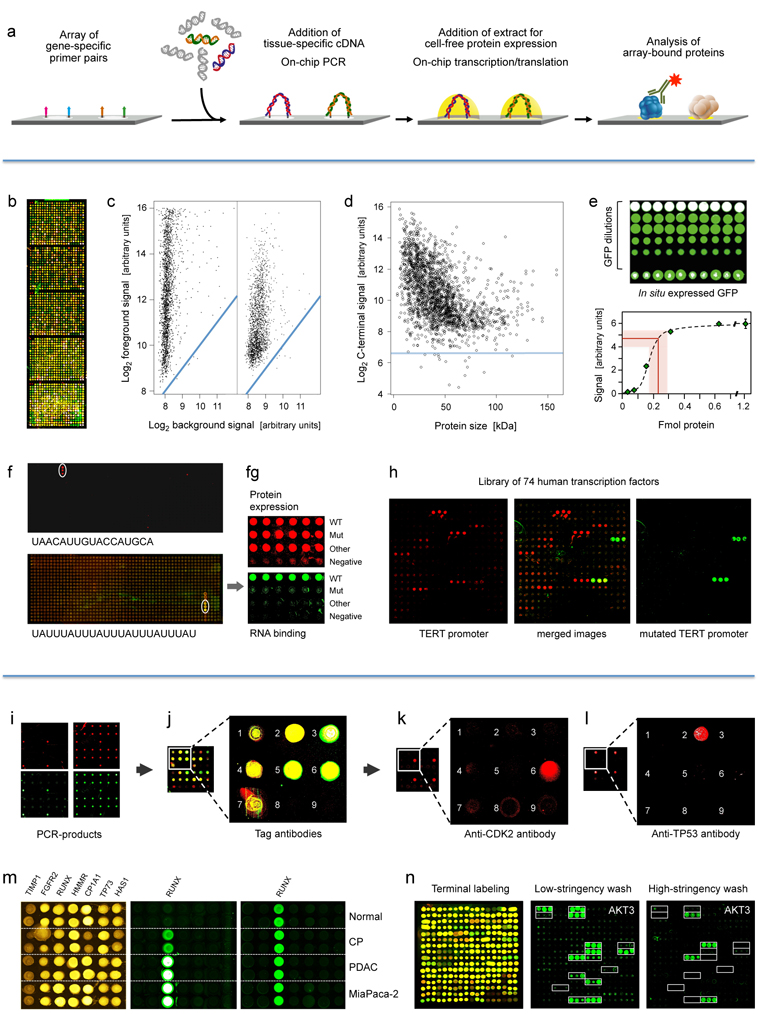
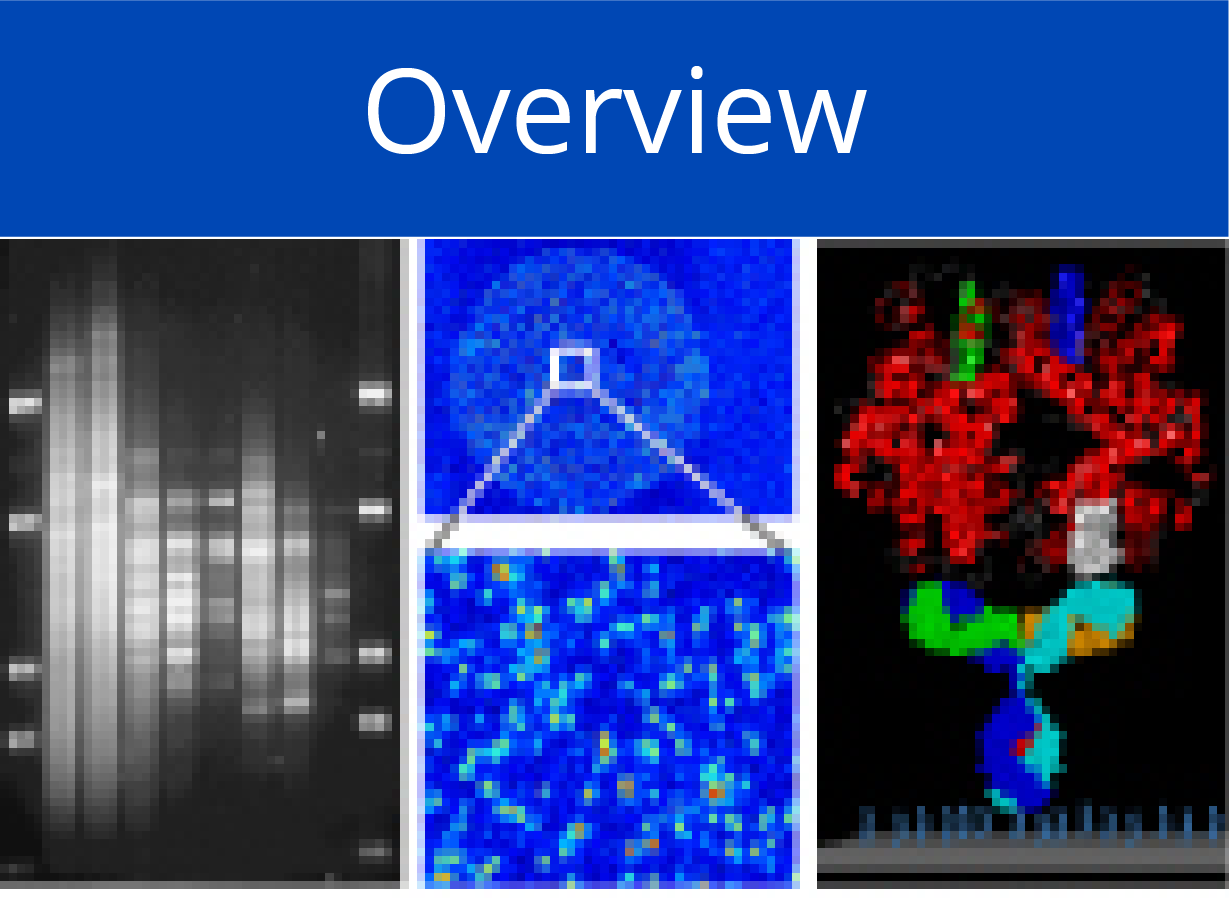
First panel: Principle and performance of the process of producing personalised protein microarrays. (a) Schematic illustration of the overall process.
Second panel: Protein microarray quality. (b) Microarray with 2016 proteins that were expressed in situ; visualisation was by incubation with red and green labelled antibodies that recognise common N- or C-terminal epitopes, respectively. (c) The typical ratio is shown of foreground to background signals for the N- (left) and C-terminus (right); blue lines indicate regions of identical intensity. (d) Detection of the C-termini of the 2016 expressed proteins with Cy3-conjugated anti-V5 antibody; the horizontal line represents a signal of three standard deviations above background. (e) Determination of the amount of in situ synthesised GFP by comparison to spotted material of known concentration; twenty measurements each were done; the red line represents the average amount of synthesised protein plus/minus one standard deviation. (f) Microarrays of T. brucei proteins were incubated with the labelled, synthetic RNA sequences shown. The white circles highlight positive signals. On the lower array, also a second protein exhibited interaction. In (g), the interacting protein from the lower microarray was analysed in more detail in comparison to a derivative with one point mutation (Mut) and another, unrelated protein. The upper level shows the protein amounts, detected by antibody binding to the N-terminus. The lower panel shows binding of the synthetic RNA. Subsequent studies demonstrated a more than 3,000-fold difference in affinity to the specific RNA between wildtype and mutated protein. (h) Binding to 74 human transcription factors of a labelled synthetic DNA sequence representing the TERT promoter (left). Mutation of one base pair in the binding sequence led to a very different binding pattern (right).
Third panel: Detection of proteins generated in situ from individual samples. (i) Quality assessment of on-chip PCR by oligonucleotide hybridisation; two oligonucleotides were used, labelled red or green, respectively, each binding to one of the DNA-strands. Typical results are shown with an oligonucleotide specific to only one PCR-product present in quadruplicate (left) and a simultaneous hybridisation with oligonucleotides to all PCR-products (right). (j) Fusion of the images obtained after an incubation with fluorescently labelled antibodies against N- (red signal) and C-terminal tags (green signal) of the expressed seven tumour marker proteins. Spots 8 and 9 were negative controls without DNA-template. (k, l) Protein detection with labelled antibodies that target proteins CDK2 and TP53, respectively. (m) Results obtained on arrays produced from tissue samples of individual patients. Normal = healthy pancreas; CP = chronic pancreatitis; PDAC = pancreatic ductal adenocarcinoma; MiaPaca-2 = PDAC cell line. All proteins were identified with a tag-specific antibody (left). Binding patterns obtained with two different, isoform-specific antibodies. One isoform of the RUNX1 protein was present in all samples (right); the other one was found in diseased material only (middle). (n) Ninety-six DARPin binders were expressed in situ, each in three copies. Tag-specific antibodies identified all binders (left). The other two panels (middle, right) show binding patterns obtained upon incubation with protein AKT3. The white frames indicate the 16 binders that were expected to interact with AKT3. Different washing stringency produced distinct variations in the binding patterns.
© dkfz.deCombinatorial peptide synthesis with laser-based transfer of monomers

Nine microarrays containing proteins expressed from 5 pg each of some 14,000 PCR-products. The microarray-bound proteins were labelled with luminescent dye for quality control purposes
© dkfz.de
Laser writing is used to structure surfaces in many different ways in materials and life sciences. However, combinatorial patterning applications are still limited. In a collaborative project coordinated by colleagues at KIT, a method was developed for cost-efficient combinatorial synthesis of very-high-density peptide arrays with natural and synthetic monomers. A laser automatically transfers nanometre-thin solid material spots from different donor slides to an acceptor. Each donor bears a thin polymer film, embedding one type of monomer. Coupling occurs in a separate heating step, where the matrix becomes viscous and building blocks diffuse and couple to the acceptor surface. Furthermore, two material layers of activation reagents and amino acids can be deposited consecutively. Subsequent heat-induced mixing facilitates an in situ activation and coupling of the monomers. This allows to incorporate building blocks with click chemistry compatibility or a large variety of commercially available non-activated, for example, post-translationally modified building blocks into the array's peptides with >17,000 spots per square centimetre.
Production of high-density protein-microarrays by cell-free in situ expression
Due to the success of DNA-microarrays and the growing numbers of available protein expression clones, protein microarrays become more and more popular for the high-throughput screening of protein interactions. However, the widespread applicability of protein microarrays for this and other applications is hampered by the large effort associated with their production. Beside the requirement for a protein expression library, the actual protein expression and purification represents bottleneck.
As part of the EU-funded MolTools project, we established a process that allows the generation of protein microarrays from process by which proteins are expressed from unbound DNA template molecules on the microarray surface (or on any solid support). It comprises the spotting of DNA templates onto the surface and the transfer of a cell-free transcription and translation mix on top of the same spot in a second spotting run. Using wildtype GFP as a model protein, we demonstrated the time and template dependence of this coupled transcription and translation and showed that enough protein is produced to yield signals that are comparable to 300 µg/ml of spotted protein. Plasmids as well as unpurified PCR-products can be used as templates and as little as 35 fg of PCR-product (~22,500 molecules) are sufficient for the expression of full-length proteins.
We adapted the system to the high-throughput expression of libraries by designing a single primer pair harbouring promoter, ribosomal binding site and terminator sequences for an on the chip expression of a multitude of such PCR-products. Utilising full-length cDNA libraries of overall 16,000 human clones, we are producing such microarrays for various types of analysis.
Utilising the capability of detecting the interaction of individual molecules and therefore being able to count the actual number of interacting molecules, we are able to perform interaction studies in a really quantitative manner.