Antibody Microarrays
- The impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells
- The structural basis of nanobody unfolding reversibility and thermoresistance
- Comparison of the tumour cell secretome and patient sera for an accurate serum-based diagnosis of pancratic ductal adenocarcinoma
- Detailed protocols for expression profiling by antibody microarrays
- Affinomics: Proteome binders for characterisation of human proteome function; generation, validation, application

The impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells
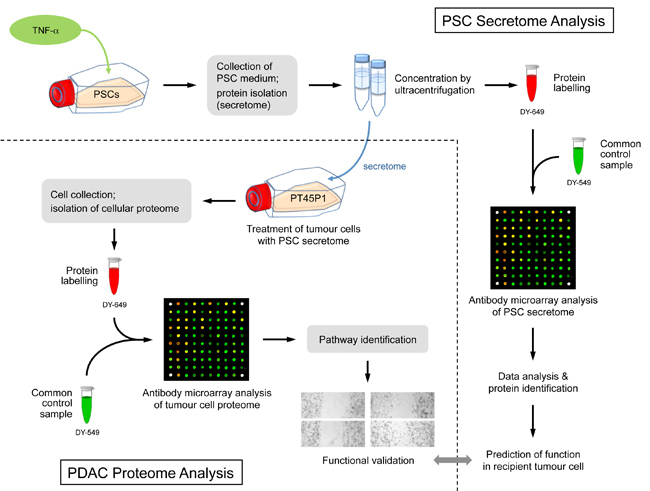
Figure legend: Scheme of the overall experimental set-up. First, the protein content of the secretome of activated PSCs was analysed and predictions were made about the functional consequences, which the secreted proteins would have in recipient cells. Second, tumour cells were grown in media conditioned with secretome. The intracellular proteome was studied and used for functional predictions. The predictions from secretome and intracellular proteome were compared and validated by investigating the actual functional variations observed and by identifying relevant regulative factors.
© dkfz.de
Pancreatic ductal adenocarcinoma (PDAC) exists in a complex desmoplastic microenvironment. As part of it, pancreatic stellate cells (PSCs) provide a fibrotic niche, stimulated by a dynamic communication between activated PSCs and tumour cells. Investigating how PSCs contribute to tumour development and for identifying proteins that the cells secrete during cancer progression, we studied by means of complex antibody microarrays the secretome of activated PSCs. A large number of secretome proteins were associated with cancer-related functions, such as cell apoptosis, cellular growth, proliferation and metastasis.
Their effect on tumour cells could be confirmed by growing tumour cells in medium conditioned with activated PSC secretome. Analyses of the tumour cells proteome and mRNA revealed a strong inhibition of tumour cell apoptosis, but promotion of proliferation and migration.
Many cellular proteins that exhibited variations were found to be under the regulatory control of eukaryotic translation initiation factor 4E (eIF4E), whose expression was triggered in tumour cells grown in the secretome of activated PSCs. Inhibition by an eIF4E siRNA blocked the effect, inhibiting tumour cell growth in vitro.
Our findings show that activated PSCs acquire a pro-inflammatory phenotype and secret proteins that stimulate pancreatic cancer growth in an eIF4E-dependent manner, providing further insight into the role of stromal cells in pancreatic carcinogenesis and cancer progression.
Publications:
Marzoq et al. (2019) Sci. Rep. 9, 5303. [PDF]
The structural basis of nanobody unfolding reversibility and thermoresistance
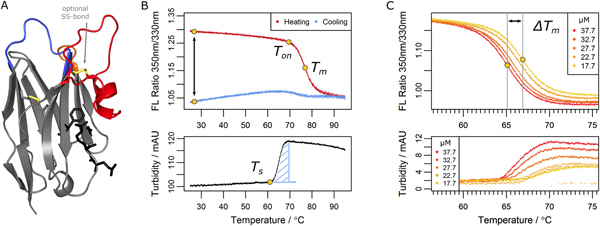
Figure legend: Parameters determined in the nanobody analysis. (A) A typical nanobody scaffold is shown. CDR loops are highlighted: CDR1, blue; CDR2, orange; CDR3, red. Hallmark positions are shown as black sticks, conserved and optional disulfide bonds as yellow sticks. (B & C) Parameters obtained from differential scanning flourimetry and turbidity assays at a temperature range of 25°C to 95°C. (B) Upper panel: The ratio of intrinsic protein fluorescence emission (350 nm/330 nm) reports about the onset temperature of unfolding (Ton) and the melting point (Tm) during the heating phase. A difference of zero between initial and final ratio values after a complete temperature cycle (black arrow) would indicate complete reversibility. Lower panel: The turbidity trace of the heating phase yields the onset temperature of aggregation (Ts) and the turbidity integral (blue shaded area); the latter serves as a qualitative measure of aggregation. If Ts occurs during the cooling phase, the turbidity integral is determined in reverse orientation. (C) Upper panel: Apparent melting temperature (Tm) values yield the ΔTm shift when aggregation is modulated by the nanobody concentration. The ΔTm shift can serve as a measure of aggregation propensity. Lower panel: the directly related turbidity traces are shown.
© dkfz.de
Nanobodies represent the variable binding domain of camelid heavy-chain antibodies and are employed in a rapidly growing range of applications in biotechnology and biomedicine. Their success is based on unique properties including their assumed ability to reversibly refold after denaturation. By characterizing nearly 70 nanobodies, we show that, opposed to common assumption, irreversible aggregation does occur for many binders upon heat denaturation, potentially affecting application-relevant parameters like stability, affinity and immunogenicity. However, by deriving aggregation propensities from apparent melting temperatures, we show that an optional disulfide bond suppresses nanobody aggregation. This effect is further enhanced by increasing the length of a complementarity determining loop which, although expected to destabilize, contributes to nanobody stability. The effect of such variations depends on environmental conditions, however. Nanobodies with two disulfide bonds, for example, are prone to lose their functionality in the cytosol. Our study suggests strategies to engineer nanobodies that exhibit optimal performance parameters and gives insights into general mechanisms which evolved to prevent protein aggregation.
Publications:
Kunz et al. (2018) Sci. Rep. 8, 7934. [PDF]
Kunz et al. (2017) BBA-Gen. Subjects 1861, 2196-2205. [PDF]
Comparison of the tumour cell secretome and patient sera for an accurate serum-based diagnosis of pancratic ductal adenocarcinoma
© dkfz.de
Pancreatic cancer is the currently most lethal malignancy. Toward an accurate diagnosis of the disease in body liquids, we studied the protein composition of the secretomes of 16 primary and established cell lines of pancreatic ductal adenocarcinoma (PDAC). Compared to the secretome of non-tumorous cells, 112 proteins exhibited significantly different abundances. Functionally, the proteins were associated with PDAC features, such as decreased apoptosis, better cell survival and immune cell regulation.
The result was compared to profiles obtained from 164 serum samples from two independent cohorts a training and a test set of patients with PDAC or chronic pancreatitis and healthy donors. Eight of the 112 secretome proteins exhibited similar variations in their abundance in the serum profile specific for PDAC patients, which was composed of altogether 189 proteins.
The 8 markers shared by secretome and serum yielded a 95.1% accuracy of distinguishing PDAC from healthy in a Receiver Operating Characteristic curve analysis, while any number of serum-only markers produced substantially less accurate results. Utility of the identified markers was confirmed by classical enzyme linked immunosorbent assays (ELISAs). The study highlights the value of cell secretome analysis as a means of defining reliable serum biomarkers.
Publications:
Mustafa et al. (2017) Oncotarget 8, 11963-11976. [PDF]
Detailed protocols for expression profiling by antibody microarrays
© dkfz.de
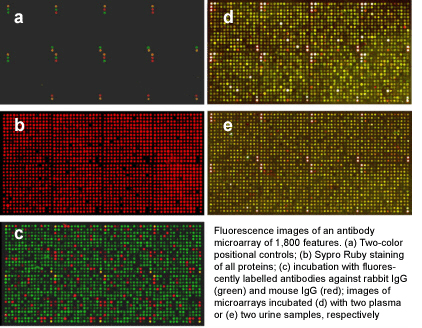
As a multiplexing technique, antibody microarrays facilitate the highly parallel detection of hundreds of different analytes from very small sample volumes of only few microliters. This is combined with a high sensitivity in the picomolar to femtomolar range, which is similar to the sensitivity of ELISA, the gold standard for protein quantification. In order to obtain such sensitivities in a robust and reproducible manner for sets of several hundreds of analytes, it is essential to optimise the experimental layout, sample handling, labelling and incubation as well as data processing steps.
Based on earlier work, we continuously developed the processing of microarrays and protein samples. In the publications listed below, we present in detail our antibody microarray protocols for multiplexed expression profiling studies, which permit the analysis of the abundance of very many proteins in plasma, urine, cell and tissue samples.
Publications:
Schröder et al. (2010) Antibody Engineering, Vol. 2, SpringerVerlag, 429-445. [PDF]
Schröder et al. (2010) Mol. Cell. Prot. 9, 1271-1280. [PDF]
Alhamdani et al. (2010) Proteomics 10, 3203-3207. [PDF]
Schröder et al. (2011) Protein Micoarrays - Meth. Mol. Biol., Springer, 203-221.
Affinomics: Proteome binders for characterisation of human proteome function; generation, validation, application
The Affinomics programme aims to leverage existing efforts in Europe to generate large-scale resources of validated protein-binding molecules (binders) as affinity reagents for characterisation of the human proteome and to apply them in comprehensive structural and functional analyses of protein expression, interactions and complexes. The project was preceded by the ProteomeBinders and AffinityProteome consortia.
Proteome targets will be focused on five categories of inter-related human proteins involved in signal transduction, cell regulation and cancer, namely protein kinases, SH2 domain-containing proteins, protein tyrosine phosphatases, proteins somatically mutated in cancers and candidate cancer biomarkers. Binders to about 1000 protein targets will be made over the course of the programme.
A high throughput, coordinated production pipeline for antigens and binders will be established. Target antigens will be expressed in three forms, as folded full-length proteins or domains, as large peptide fragments (PrESTs) based on low homology to other human proteins and as small peptides, in some cases phosphorylated. Binder types to be generated include affinity-purified polyclonal antibodies, monoclonal antibodies, recombinant antibody fragments and non-immunoglobulin scaffolds.
An important aspect will be the development of highly efficient next generation recombinant selection methods, based on phage, cell and ribosome display, capable of producing high quality binders at greater throughput and lower cost than hitherto. Systems and procedures for thorough binder validation and quality control will be established. The affinity reagents will be applied in advanced innovative and sensitive technologies for specific detection of target proteins and interacting protein complexes in cells, tissues and fluids, for improved understanding of protein function and new classes of diagnostic assays.
For more detailed information, click on the map of the consortium.
Sequence validation of binders
Antibodies and other binder types are crucial for any proteome analysis. Although many tens of thousands of antibodies are available from commercial sources and academic institutions(see antibodypedia, for example), quality and reproducibility of analyses performed with these molecules vary substantially. Antibodies exhibit huge differences in specificity and affinity, including binders that target the same protein. Worse, even binders that are supposedly from the same source showed significant lot-to-lot variation.
One reason for variation could be the actual assay; an antibody perfoming very well on Western blots may not meet the quality standards of pull-down experiments or microarray analyses, and vice versa. Another point, however, that is contributing significantly to low reproducibility is a lack of commonly accepted performance parameters and tests for their definition This is made worse by the fact that currently most binders are ill-described. Consequently, one cannot be sure, if a binder is exactly the molecule that was used in assays before.
Initiated by Andrew Bradbury, 101 scientists of the Affinomics consortium and beyond have had some thoughts about the matter. A simple solution to the problem of how to make sure that everybody is really using the very same binder molecules in their experiments would be a sequence verification of each binder. While relatively easily achievable for recombinant binders and monoclonal antibodies, it is more difficult to establish for polyclonal binders, for example. A more important obstacle for an implementation of such a scheme, however, could be the reluctance of antibody producers to share the sequences of their molecules, since this would make available their good binders to the entire scientific (and commercial) community for free.
Publications:
Bradbury et al. (2015) Nature 518, 27-29. [PDF]