Melanoma & Other Cancers
- Melanoma microRNA trafficking controls tumour primary niche formation
- Functional screens by means of lentiviral shRNA libraries
- An in vivo RNAi screen identifies SALL1 as a tumour suppressor in human breast cancer with a role in CDH1 regulation
- A mammosphere formation RNAi screen reveals genes that promote a breast cancer stem-like phenotype

Melanoma microRNA trafficking controls tumour primary niche formation

© dkfz.de
Melanoma originates in the epidermis and becomes metastatic after invasion into the dermis. Prior interactions between melanoma cells and dermis are poorly studied. Here, we show that melanoma cells directly affect the formation of the dermal tumour niche by microRNA trafficking before invasion.
Melanocytes, cells of melanoma origin, are specialized in releasing pigment vesicles, termed melanosomes. In melanoma in situ, we found melanosome markers in distal fibroblasts before melanoma invasion. The melanosomes carry microRNAs into primary fibroblasts triggering changes, including increased proliferation, migration and pro-inflammatory gene expression, all known features of cancer-associated fibroblasts (CAFs). Specifically, melanosomal microRNA-211 directly targets IGF2R and leads to MAPK signalling activation, which reciprocally encourages melanoma growth. Melanosome release inhibitor prevented CAF formation. Since the first interaction of melanoma cells with blood vessels occurs in the dermis, our data suggest an opportunity to block melanoma invasion by preventing the formation of the dermal tumour niche.
Publications:
Dror et al. (2016) Nature Cell Biol. 18, 1006-1017 [PDF]

Functional screens by means of lentiviral shRNA libraries
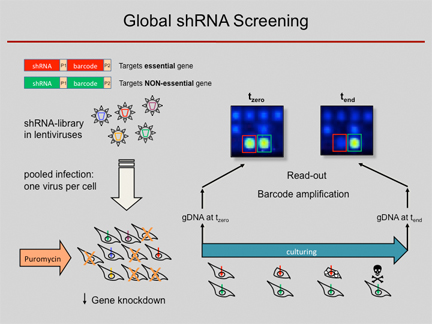
RNA interference (RNAi) has become a popular and important tool for the analysis of gene function. Loss-of-function studies, commonly performed by transfection of short interfering RNAs (siRNAs), have greatly facilitated functional analyses of the human transcriptome. However, there are major downsides to siRNA experiments, most importantly the transient inhibition of gene expression as well as their inefficient transfection into non-dividing cells. Overcoming those limitations, short hairpin RNA (shRNA) expression vectors are available, which stably integrate into a target cell's genome via retro- or lentiviral gene transfer. Intracellular processing of shRNAs results in short duplex RNAs with siRNA-like properties. Viral integration ensures a broad range of infectable target cell types and a stable expression of specific shRNAs, resulting in the permanent reduction of the targeted gene product. Complex shRNA expression libraries allow the targeted knockdown of thousands of different genes in a single experiment.
Using such lentiviral vector shRNA libraries and initially barcode arrays and meanwhile next-generation sequencing analysis for decoding of the pooled RNAi screens, we are able to quantify the abundance of individual shRNAs and thus determine in a complex pool the number of cells infected with an individual shRNA construct. We used the technique to predict anti-proliferative effects of individual shRNAs from pooled negative selection screens, for example, identified synthetic-lethal activities toward combination therapies, defined genes which are required for a stem-cell like phenotype and found tumour suppressor genes by in vivo studies. For more details, see below.
Further studies are under way, both for the elucidation of basic regulative processes associated to cancer and for the identification of pathways that are affected by particular drugs or compounds. In particular, we use the technique for obtaining more detailed information on the functional effects of particularly potentially druggable gene products.
More recently, CRISPR-Cas constructs have been used for similar analyses and other purposes.
An in vivo RNAi screen identifies SALL1 as a tumour suppressor in human breast cancer with a role in CDH1 regulation
The gold standard for determining the tumorigenic potential of human cancer cells is a xenotransplantation into immunodeficient mice. Higher tumorigenicity of cells is associated with earlier tumour onset.
We employed xenotransplantation to assess the tumorigenic potential of human breast cancer cells following RNAi-mediated inhibition of over 5,000 genes. We identified sixteen candidate tumor suppressors, one of which is the zinc finger transcription factor SALL1. Analysing this particular molecule in more detail, we showed that inhibition of SALL1 correlated with reduced levels of CDH1, an important contributor to epithelial-to-mesenchymal (EMT) transition. Furthermore, SALL1 expression led to increased migration and more than twice as many cells expressing a cancer stem cell signature. Also, SALL1 expression correlated with the survival of breast cancer patients. These findings cast new light on a gene, which has previously been described to be relevant during embryogenesis, but not carcinogenesis.
Publications:
Wolf et al. (2014) Oncogene 33, 4273-4278. [PDF]
A mammosphere formation RNAi screen reveals genes that promote a breast cancer stem-like phenotype
Breast cancer stem cells are suspected to be responsible for tumour recurrence, metastasis formation as well as chemoresistance. Consequently, great efforts are made to understand the molecular mechanisms underlying cancer stem cell maintenance. In order to study these rare cells in vitro, they are typically enriched via mammosphere culture. We developed a mammosphere-based negative selection shRNAi screening system suitable to analyse the involvement of thousands of genes in the survival of cells with cancer stem cell properties. We used a sub-population with cancer stem cell properties of cell line SUM149 that were enriched in mammospheres. Identified candidates were validated in mammosphere and soft agar colony formation assays. Further, we evaluated the influence of their expression on the stem cell sub-population. Also, the tumorigenic potential of SUM149 after up- or down-regulation of candidates was examined by xenograft experiments.
Using this approach, Jak-STAT as well as cytokine signalling were identified to be involved in mammosphere formation. Furthermore, the autophagy regulator ATG4A was found to be essential for maintenance of the cancer stem cell sub-population and regulation of breast cancer cell tumourigenicity in vivo.
Publications:
Wolf et al. (2013) Breast Cancer Res. 15, R109. [PDF]