Epigenetics
- DNA-methylation studies
- M6-Methyladenoadenosine landscape of glioma stem-like cells: METTL3 is essential for the expression of actively transcribed genes and sustenance of the oncogenic signalling
- Methylation in the gene encoding the long intergenic non-coding RNA 299 serves as biomarker in peripheral blood for triple-negative breast cancer
- GHSR DNA hypermethylation: a common epigenetic alteration of high diagnostic value in many cancers
- Early epigenetic down-regulation of microRNA-192 expression promotes pancreatic cancer progression

DNA-methylation studies
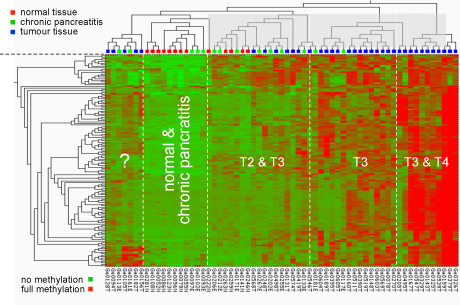
Methylation pattern at particular CpG dimers in the promoters of cancer-relevant genes. The type of pancreas tissue analysed is shown at the top; the degree of methylation is indicated by a colour-code as shown at the bottom, ranging from green (no methylation) to red (hypermethylation).
© dkfz.de
Changes in genomic DNA methylation patterns are one of the earliest and most consistent features of tumourigenesis. It has been demonstrated that aberrant DNA methylation profiles can be used as a valuable markers for clinical tumour characterisation. Technically, we initially applied microarray technology toward a genome-wide and high-resolution analysis of DNA methylation patterns. Meanwhile, next-generation sequencing and other techniques have replaced the arrays entirely.
The detection of methylation variations is mostly performed by using bisulfite treatment to uncover the methylation status. Sodium bisulfite induces methylation-dependent single-nucleotide polymorphisms by converting unmethylated cytosine to uracil and, upon PCR amplification, to thymine (see figure below). 5-Methylcytosine is not affected by sodium bisulfite treatment and thus amplified as cytosine. The conversion can be identified by any means of sequence analysis.

The data are evaluated in combination with available clinical data and information from other analyses, such as transcript profiling. This could allow fundamental insights into the role of DNA methylation during tumourigenesis. We study in some detail the functional consequences of variations of particularly promoter methylation, with a focus on promoters of microRNA genes, and have been successful with uncovering relevant functional mechanisms in several cancer entities.
M6-Methyladenoadenosine landscape of glioma stem-like cells: METTL3 is essential for the expression of actively transcribed genes and sustenance of the oncogenic signalling
Despite recent advances in m6A biology, the regulation of crucial RNA processing steps by the RNA methylase METTL3 in glioma stem-like cells (GSCs) remains obscure. An integrated analysis of m6A-RNA-immunoprecipitation and total RNA-sequencing of METTL3-silenced GSCs identified that m6A modification in GSCs is principally carried out by METTL3. The m6A-modified transcripts showed higher abundance compared to non-modified transcripts. Further, METTL3 is essential for the expression of GSC-specific actively transcribed genes. Silencing METTL3 resulted in an elevation of several aberrant, alternative splicing events. Putative m6A reader proteins play a key role in the RNA stabilization function of METTL3. METTL3 altered A-to-I and C-to-U RNA editing events by differentially regulating RNA editing enzymes ADAR and APOBEC3A. Similar to protein-encoding genes, lincRNAs with m6A-marks showed METTL3-dependent high expression. m6A modification of 3-UTRs appears to result in a conformation-dependent hindrance of miRNA binding to their targets. The integrated analysis of the m6A regulome in METTL3-silenced GSCs showed global disruption in tumorigenic pathways that are indispensable for GSC maintenance and glioma progression. We conclude that METTL3 plays a vital role in many steps of RNA processing and orchestrates successful execution of oncogenic pathways in GSCs.
Publications:
Visvanathan et al. (2019) Genes 10, 141.
Methylation in the gene encoding the long intergenic non-coding RNA 299 serves as biomarker in peripheral blood for triple-negative breast cancer

Manhattan plot showing the chromosomal distribution of the p-values of 370,706 methylation variations in TNBC. The dashed line indicates the 0.001 significance threshold.
© dkfz.de
Worldwide, breast cancer was the most frequently diagnosed cancer and cause of cancer death among women in 2012. Triple-negative breast cancer (TNBC), defined by lack of (i) tumour expression of estrogen receptor (ER), (ii) progesterone receptor (PR), and (iii) human epidermal growth factor receptor 2 (HER2), is the most aggressive subtype of breast cancer and accounts for 10% to 20% of all diagnoses. We screened the epigenome-wide methylation profiles of 233 TNBC patients and 233 age-matched controls (discovery set) and validated the most promising methylation probes in 57 TNBC patients and 124 controls (validation set) with the aim of identifying epigenetic biomarkers based on DNA from peripheral blood leukocytes.
Publications:
Bermejo et al. (2019) Epigenomics 11, 81.
GHSR DNA hypermethylation: a common epigenetic alteration of high diagnostic value in many cancers
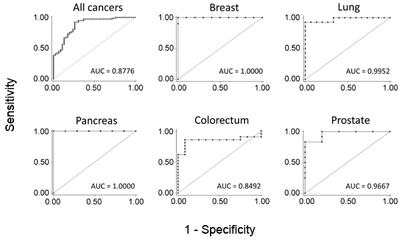
Typical ROC diagrames are shown for five particular cancer entities. Also the overall accuracy for all studied cancers is presented. The AUC value indicates the respetive degree of diagnostic accuracy.
© dkfz.de
Diagnosis
Identification of a single molecular trait that is determinant of common malignancies may serve as a powerful diagnostic supplement to cancer type-specific markers. Substantial hypermethylation at the promoter and first exon of growth hormone secretagouge receptor (GHSR) was found to be characteristic of seven studied malignancies with very high sensitivity and specificity. Discrimination of breast or pancreatic cancer from healthy tissue samples exhibited 100% specificity and sensitivity, for example.
Early detection
Moreover, differential methylation was observed for ductal carcinoma in situ (DCIS), for instance, which is considered an early stage breast cancer that may progress to invasive cancer. GHSR gene methylation could therefore be particularly attractive for early detection, which is a key factor in cancer control.
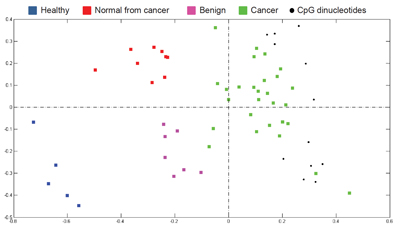
To visualise differences in the degree of methylation in breast samples, correspondence analysis (CA) was used. In the projection plot, each sample is depicted as a coloured square and CpG sites that exhibited the most significant differential methylation levels are represented as black dots. All co-localise with the cancer samples at the right side of the plot, indicating that the highest methylation level is found in cancer. In contrast, the healthy samples are located to the left, in the opposite direction off the centroid, indicating that the CpGs are at the lowest level of methylation in these samples. Likewise, based on the localisation of normal-appearing tissues of cancer patients and benign samples along the horizontal axis (first principal component; i.e., the direction along which the samples show the largest variation), it can be seen that an intermediate methylation load existed in these samples
© dkfz.de
Field defect in sporadic cancers
The increased methylation of the signature CpGs was also present in normal-appearing tissues collected from cancer patients, but not in samples obtained from healthy donors. This suggests the involvement of DNA methylation in a field defect. Field defect (or field cancerization) refers clinically to the existence of pre-neoplastic alterations in cells of a tissue that are associated with local recurrences. From a molecular point of view, this phenomenon has been explained by genetic abnormalities in patients with familial cancers. However, our data propose a contribution of epigenetic alterations to the field defect in sporadic cancers. GHSR methylation could thus possibly act as a marker of treatment success and recurrence.
Publications:
Amini et al. (2019) J. Cell. Physiol., in press. [PDF]
Jandaghi et al. (2015) Cell Cycle 14, 689. [PDF]
Moskalev et al. (2015) Oncotarget 6, 4418. [PDF]
Botla et al. (2012) Breat Cancer Res. Treat. 136, 705. [PDF]
Early epigenetic down-regulation of microRNA-192 expression promotes pancreatic cancer progression

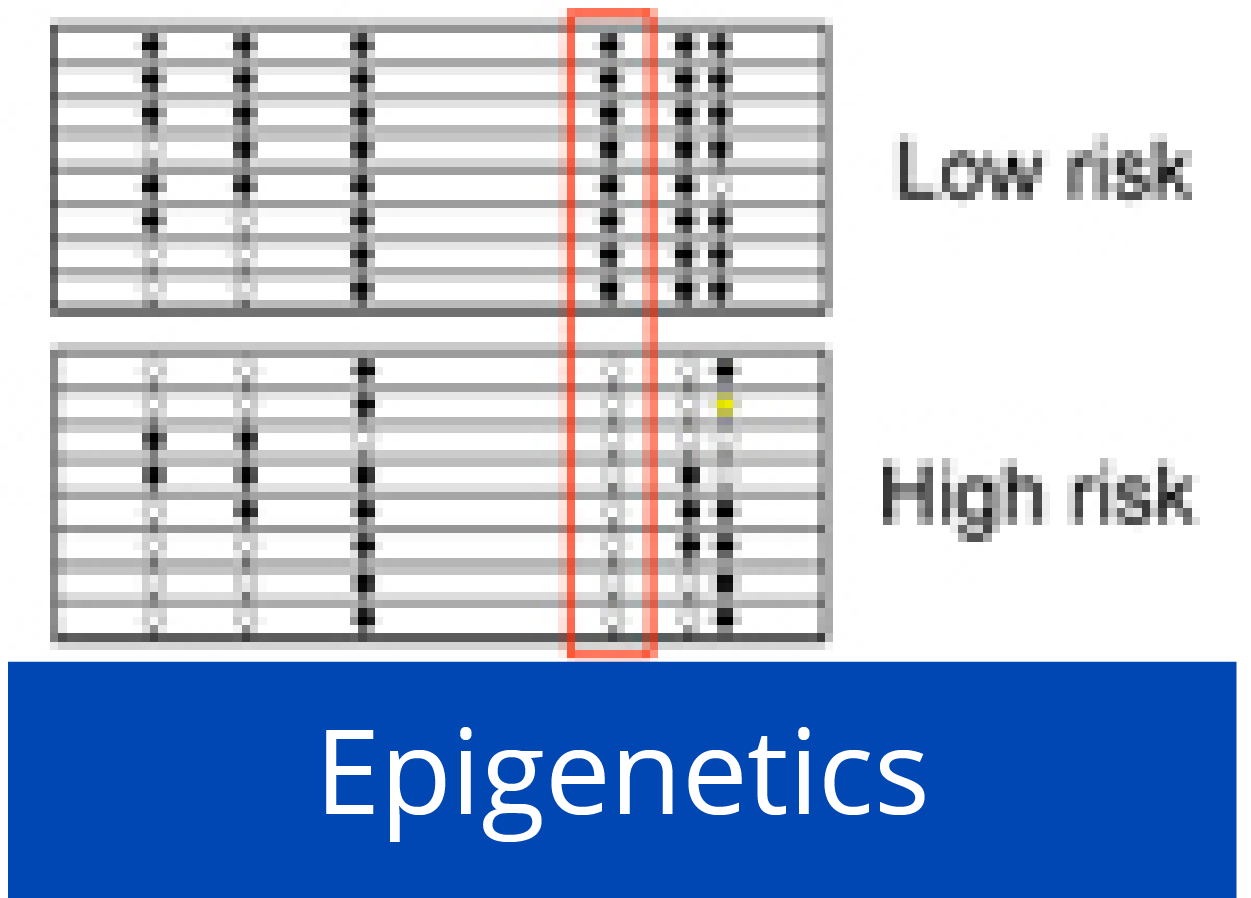
Down-regulation of miR-192 in chronic pancreatitis (CP) and PDAC patient samples compared to normal pancreas (NP) is epigenetically regulated. (left) Pooled bisulfite sequencing of the CpG island located 3.5 kb upstream of miR-192 revealed an increase in its methylation status in PDAC and CP samples compared to NP (boxed region in the heatmap). (right) Hypermethylation inversely correlated with expression of miR-192 in CP and PDAC.
© dkfz.de
Pancreatic ductal adenocarcinoma (PDAC) is characterized by very early metastasis, suggesting the hypothesis that metastasis-associated changes may occur prior to actual tumor formation. We identified miR-192 as an epigenetically regulated suppressor gene with predictive value in this disease. miR-192 was downregulated by promoter methylation in both PDAC and chronic pancreatitis (CP), the latter of which is a major risk factor for development of PDAC. Functional studies in vitro and in vivo in mouse models of PDAC showed that overexpression of miR-192 was sufficient to reduce cell proliferation and invasion. Mechanistic analyses correlated changes in miR-192 promoter methylation and expression with epithelial-mesenchymal transition (EMT). Cell proliferation and invasion were linked to altered expression of the miR-192 target gene SERPINE1 that is encoding the protein plasminogen activator inhibitor-1 (PAI-1), an established regulator of these properties in PDAC cells. Notably, our data suggested that invasive capacity was altered even before neoplastic transformation occurred, as triggered by miR-192 downregulation. Overall, our results highlighted a role for miR-192 in explaining the early metastatic behavior of PDAC and suggested its relevance as a target to develop for early diagnostics and therapy.
Publications:
Botla et al. (2016) Cancer Res. 76, 4149. [PDF]