Research
- Research Topics
- Cell Biology and Tumor Biology
- Stem Cells and Cancer
- Inflammatory Stress in Stem Cells
- Experimental Hematology
- Molecular Embryology
- Signal Transduction and Growth Control
- Epigenetics
- Redox Regulation
- Vascular Oncology and Metastasis
- Clinical Neurobiology
- Molecular Neurogenetics
- Molecular Neurobiology
- Mechanisms Regulating Gene Expression
- Molecular Biology of Centrosomes and Cilia
- Dermato-Oncology
- Pediatric Leukemia
- Tumour Metabolism and Microenvironment
- Personalized Medical Oncology
- Molecular Hematology - Oncology
- Cancer Progression and Metastasis
- Translational Surgical Oncology
- Neuronal Signaling and Morphogenesis
- Cell Signaling and Metabolism
- Cell Fate Engineering and Disease Modeling
- Cancer Drug Development
- Cell Morphogenesis and Signal Transduction
- Functional and Structural Genomics
- Molecular Genome Analysis
- Molecular Genetics
- Pediatric Neurooncology
- Cancer Genome Research
- Chromatin Networks
- Functional Genome Analysis
- Theoretical Systems Biology
- Neuroblastoma Genomics
- Signaling and Functional Genomics
- Signal Transduction in Cancer and Metabolism
- RNA-Protein Complexes and Cell Proliferation
- Systems Biology of Signal Transduction
- Areas of Interest
- Advancement of clinical proteomics for systems medicine
- Bridging from the single cell to the cell population Epo-induced cellular responses and erythroleukemia
- Deciphering tumor microenvironment interactions determining lung cancer development
- Mechanisms controlling the compensation of liver injury and towards model-based biomarkers for early detection of liver cancer
- Application of dynamic pathway modelling for personalized medicine
- Group Members
- Publications
- Open Positions
- Funding
- Teaching
- Areas of Interest
- Molecular thoracic Oncology
- Proteomics of Stem Cells and Cancer
- Computational Genomics and System Genetics
- Applied Functional Genomics
- Applied Bioinformatics
- Translational Medical Oncology
- Metabolic crosstalk in cancer
- Pediatric Glioma Research
- Cancer Epigenomics
- Translational Pediatric Sarcoma Research
- Artificial Intelligence in Oncology
- Mechanisms of Genomic Variation and Data Science
- Neuropathology
- Pediatric Oncology
- Neurooncology
- Somatic Evolution and Early Detection
- Translational Control and Metabolism
- Soft-Tissue Sarcoma
- Precision Sarcoma Research
- Brain Mosaicism and Tumorigenesis
- Mechanisms of Genome Control
- Translational Gastrointestinal Oncology and Preclinical Models
- Translational Lymphoma Research
- Mechanisms of Leukemogenesis
- Genome Instability in Tumors
- Developmental Origins of Pediatric Cancer
- Brain Tumor Translational Targets
- Translational Functional Cancer Genomics
- Regulatory Genomics and Cancer Evolution
- SPRINT
- Cancer Risk Factors and Prevention
- Cancer Epidemiology
- Biostatistics
- Clinical Epidemiology and Aging Research
- Health Economics
- Physical Activity, Prevention and Cancer
- Preventive Oncology
- Personalized Early Detection of Prostate Cancer
- Digital prevention, diagnostics and therapy guidance
- Tumorigenesis and molecular cancer prevention
- Genomic Epidemiology
- Cancer Survivorship
- Immunology, Infection and Cancer
- Structural Biology of Infection and Immunity
- Cellular Immunology
- B Cell Immunology
- Immune Diversity
- Immunoproteomics
- Personalized Immunotherapy
- mRNA Cancer Immunotherapies
- Tumor Immunology and Tumor Immunotherapy
- Infections and Cancer Epidemiology
- Pathogenesis of Virus-Associated Tumors
- Immunotherapy and Immunoprevention
- Virus-associated Carcinogenesis
- Chronic Inflammation and Cancer
- Microbiome and Cancer
- Molecular Oncology of Gastrointestinal Tumors
- Applied Tumor Immunity
- Neuroimmunology and Brain Tumor Immunology
- Applied Tumor Biology
- Virotherapy
- Adaptive Immunity and Lymphoma
- Dermal Oncoimmunology
- Immune Regulation in Cancer
- Systems Immunology and Single Cell Biology
- Pediatric Immuno-Oncology
- Epithelium Microbiome lnteractions
- Experimental Hepatology, Inflammation and Cancer
- GMP & T Cell Therapy
- Tumorvirus-specific Vaccination Strategies
- Mammalian Cell Cycle Control Mechanisms
- Molecular Therapy of Virus-Associated Cancers
- DNA Vectors
- Episomal-Persistent DNA in Cancer- and Chronic Diseases
- Immune Monitoring
- News
- Imaging and Radiooncology
- Radiology
- Research
- Computational Radiology Research Group
- Contrast Agents In Radiology Research Group
- Neuro-Oncologic Imaging Research Group
- Radiological Early Response Assessment Of Modern Cancer Therapies
- Imaging In Monoclonal Plasma Cell Disorders
- 7 Tesla MRI - Novel Imaging Biomarkers
- Functional Imaging
- Visualization And Forensic Imaging
- PET/MRI
- Dual- and Multienergy CT
- Radiomics Research Group
- Prostate Research Group
- Bone marrow
- Musculoskeletal Imaging
- Microstructural Imaging Research Group
- Staff
- Patients
- Research
- Medical Physics in Radiology
- X-Ray Imaging and Computed Tomography
- Federated Information Systems
- Translational Molecular Imaging
- Medical Physics in Radiation Oncology
- Biomedical Physics in Radiation Oncology
- Intelligent Medical Systems
- Medical Image Computing
- Radiooncology - Radiobiology
- Smart Technologies for Tumor Therapy
- Team
- Research
- Microrobots and Miniaturize Devices for Minimally-invasive Surgery
- Magnetic localization and sensing for biomedical devices
- Nanorobots for Targeted Delivery in Deep Biological Tissues
- 3D Additive Manufacturing of Soft Materials as In Vitro Tumor Models
- Surgical Simulation on Cyber-physical Organ Models
- News
- Vacancies
- Radiation Oncology
- Molecular Radiooncology
- Nuclear Medicine
- Translational Radiation Oncology
- Translational Radiotheranostics
- Interactive Machine Learning
- Intelligent Systems and Robotics in Urology
- Multiparametric methods for early detection of prostate cancer
- Translational Molecular Imaging in Oncologic Therapy Monitoring
- Radiology
- Cell Biology and Tumor Biology
- Research Groups A-Z
- Junior Research Groups
- Core Facilities
- News
- List of Core Facilities
- Antibodies
- Cellular Tools
- Center for Preclinical Research
- Central Library
- Chemical Biology
- Dieter Morszeck Biorepository
- Electron Microscopy
- Flow Cytometry
- Information Technology ITCF
- Light Microscopy
- Metabolomics
- Microarray
- Microbiological Diagnostics
- Next Generation Sequencing
- Omics IT and Data Management
- Proteomics
- Radiopharmaceuticals and Preclinical Trials
- Single-cell Open Lab
- Small Animal Imaging
- Transgenic Service
- Tumor Models
- OMERO@DKFZ
- List of Technologies
- DKFZ Core Facilities Publication Policy
- Enabling Technology
- Data Science @ DKFZ
- INFORM
- Baden-Württemberg Cancer Registry
- Cooperations & Networks
- National Cooperations
- International Cooperations
- Cooperational Research Program with Israel: DKFZ - MOST in Cancer Research
- Program
- Members of the Program Committee
- Call
- Publication Database
- German-Israeli Cancer Research Schools
- Archive
- Heidelberg - Israel, Science and Culture
- Symposium 40 Years of German-Israeli Cooperation
- 35th Anniversary Symposium
- 34th Meeting of the DKFZ-MOST Program
- 40th Anniversary Publication
- 30th Anniversary Publication
- 20th Anniversary Publication
- Flyer - The Cancer Cooperation Program
- List Publications 1976-2004
- Highlight-Projects
- Cooperational Research Program with Israel: DKFZ - MOST in Cancer Research
- Cooperations with industrial companies
- DKFZ PostDoc Network
- Cross Program Topic RNA@DKFZ
- Cross Program Topic Epigenetics@dkfz
- Cross Program Topic Single Cell Sequencing
- WHO Collaborating Centers
- DKFZ Site Dresden
- Health + Life Science Alliance Heidelberg Mannheim
Division of Computational Genomics and Systems Genetics
Prof. Dr. Oliver Stegle
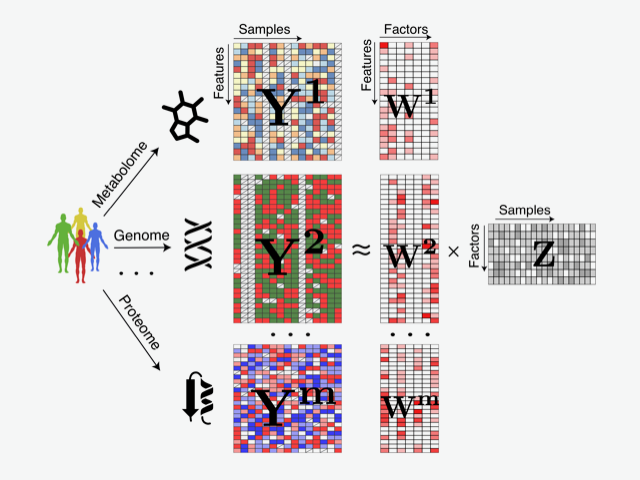
Illustration of a statistical method for integrating multiple omics datasets. Multi?Omics Factor Analysis (MOFA) is a computational framework for unsupervised discovery of the principal axes of biological and technical variation when multiple omics assays are applied to the same samples.
© dkfz.de
Our interest lies in computational methods to unravel the genotypephenotype map on a genome-wide scale. How do genetic background and environment jointly shape phenotypic traits or causes diseases? How are genetic and external factors integrated at different molecular layers, and how variable are molecular states between individual cells?
We use statistics and machine learning as our main tool to address these questions. To make accurate inferences from high-dimensional omics datasets, it is essential to account for biological and technical noise and to propagate evidence strength between different steps in the analysis. We develop methods that enable connecting genetic factors to phenotypes and to integrate multiomics data in health and disease.
Our methodological work ties in with biomedical collaborations, and we are developing methods to fully exploit high-throughput datasets from the most recent profiling technologies. In doing so, we derive computational methods to dissect phenotypic variability at the level of the transcriptome and the proteome and we derive new tools for single-cell biology.
Future Outlook
We will continue to develop innovative statistical approaches to analyse data from high-throughput genetic and molecular profiling studies. We are particularly interested in following up our recent efforts to model single-cell variation data. A major challenge in this field will be the integration of multiple data modalities in the same cells, for example linking single-cell epigenome variation with single-cell transcriptomes. We will carry out this work as an active partner in the Human Cell Atlas project (https://www.humancellatlas.org).
