Division of Functional Genome Analysis
Dr. Jörg D. Hoheisel
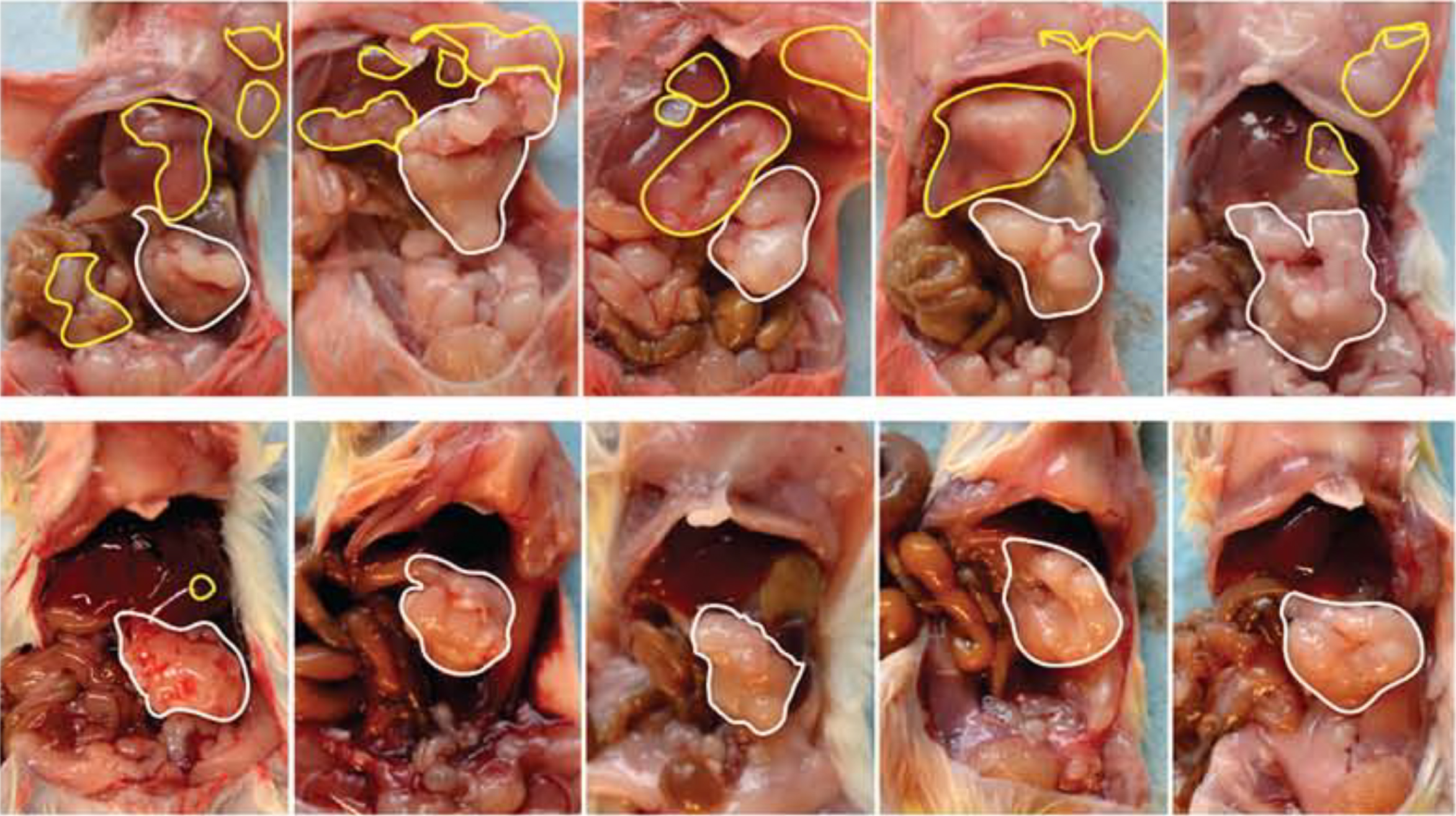
Potential of DRD2 antagonist Haloperidol a molecule already used as an antipsychotic drug as a therapeutic approach to treat pancreatic cancer. Representative images show pancreatic tumours in mice. The animals in the upper row were untreated; the bottom row shows the results obtained with Haloperidol treatment. White and yellow outlines indicate primary and metastatic tumours, respectively. Treatment with Haloperidol reduced the metastasis dissemination strongly.
© dkfz.de
Research at the Division of Functional Genome Analysis focuses on an assessment and description of the realisation and regulation of cellular function from genetic information.
Analyses of tumour material are at the centre of attention, with an emphasis on pancreatic cancer. The effect of DNA sequence variations and epigenetic modulation of the genome are studied. This is combined with measurements on variations in transcription factor binding and changes in transcript levels of coding and noncoding RNAs. Concomitantly, protein expression is analysed, mostly by means of affinity-based assay formats. We complement the molecular analyses with functional studies for the elucidation of relevant cellular mechanisms.
Beside the creation of basic scientific knowledge, we aim at establishing means for reliable, potentially early and non-invasive molecular diagnosis, which translates also into accurate prognosis, patient stratification and monitoring of treatment results. Another objective is the identification of new therapeutic modalities.
Currently, we have particular focus on studying proteomic variations. Affinity-based processes have been established that permit a robustness and reproducibility that meet the requirements of clinical applications and are amendable to translation. One scientific objective is the identification of disease-specific protein isoforms; structural variation is often an indicator of different functional activity. Also, measurements of protein interactions are performed, in particular for the identification of variations that occur at a personal level. A third major activity is the creation of a map of the protein-mediated communication between the different cell types within (pancreatic) tumour microenvironment or with the wider peritumoral tissue.
Another line of work aims at the fully synthetic, in vitro implementation of complex biological processes. Motivation is their utilization for the production of biomedically active molecules, such as non-immunogenic agents, and the establishment of an entirely artificial molecular system. Cell-free biosynthetic production will be crucial for mastering many biotechnological and pharmacochemical challenges ahead. In addition, artificial biological systems will complement Systems Biology by evaluating biological models experimentally. Similar to physics, an iterative process of performing experimental and theoretical studies will provide insight into cellular functioning. Eventually, this may lead to the establishment of a fully synthetic self-replicating system and, ultimately, an archetypical model of a cell.
