Research Projects
- I. Understanding the dynamics and regulation of cell-intrinsic antiviral signaling
- II. Involvement of cell-intrinsic antiviral signaling in tumor development and therapy
- III. Understanding the dynamics of viral replication and its interplay with the cellular antiviral system
- IV. Our Pandemic Special Investigations into the innate immune response against SARS-CoV2
I. Understanding the dynamics and regulation of cell-intrinsic antiviral signaling
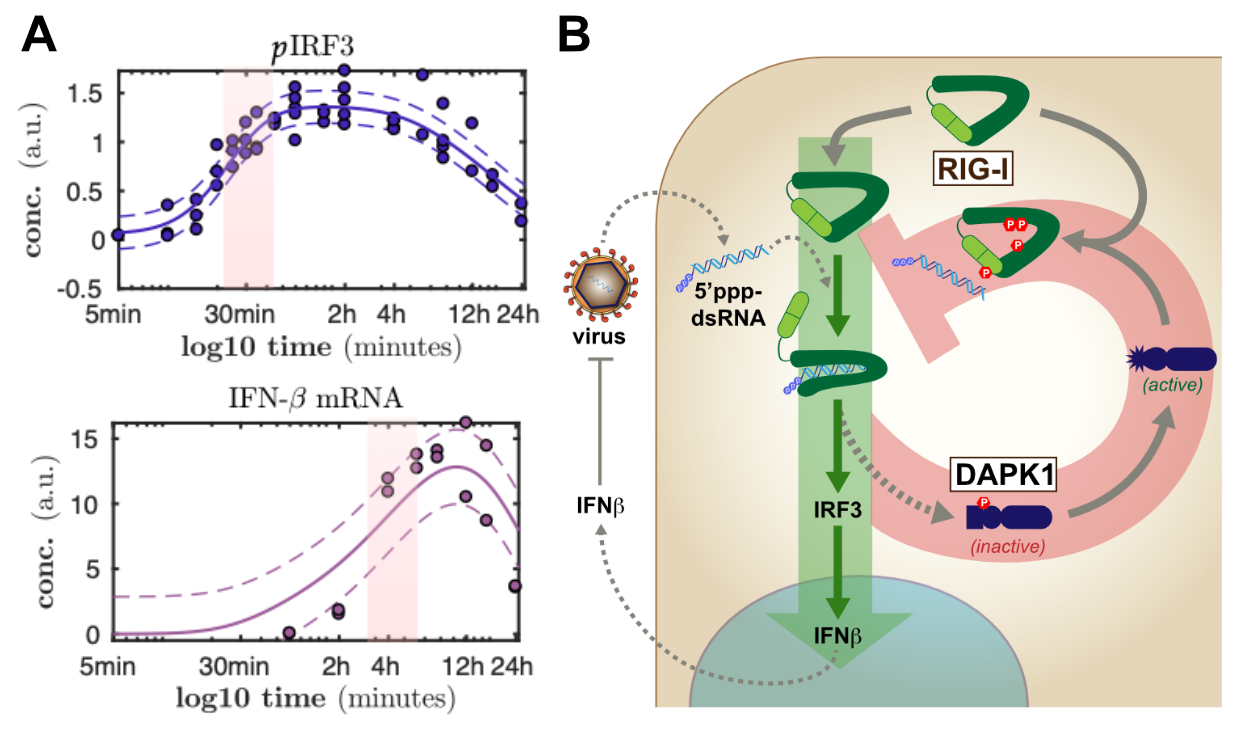
Panel (A) shows examples of temporally highly resolved measurements of activation of two different stages of antiviral signaling. Similarly, we characterized many further steps of RIG-I and interferon signaling in order to obtain a full understanding of the kinetics of the antiviral response. (B) schematically shows the regulator role of DAPK1 in the termination of RIG-I antiviral signaling in a negative feedback loop.
© dkfz.de
In order to sense a viral infection and trigger an appropriate defensive response, the cell has developed a set of sensor molecules. One of the most prominent sensors is retinoic acid inducible gene I (RIG-I). Upon recognition of viral RNA, RIG-I triggers a signaling cascade through the crucial innate adapter molecule MAVS, eventually leading to activation of the latent transcription factors IRF-3 and NFkB. These transcription factors in turn drive the production and secretion of type I and III interferons, which are highly potent mediators of the antiviral response, driving the infected as well as surrounding cells into an alert antiviral state, which, in most cases, is sufficient in restricting the replication and spread of the virus.
We are very much interested in understanding the dynamics of this vital signaling system and how it is modulated by cellular regulators as well as virus encoded antagonists. For the body, on the one hand, it is extremely important to tightly regulate these potent responses as aberrant activation or prolonged / overshooting activity of these systems are known to cause severe damage to the organism. On the other hand, upon recognition of only a tiny amount of non-self, virus-derived molecules, the defense system has fire up instantly in order to stop the fast exponential replication of the pathogen. We have established different technological approaches to study the dynamics of these processes (figure 1A) and to identify and characterize molecular regulators.
The dynamics of cell-intrinsic antiviral defense responses is crucial as it is racing against extremely fast pathogens. As viruses also fight back, frequently encoding very potent antagonists of innate immune pathways, it is vital for the cell to stay ahead. In a long and tedious project, we have recently finished the most comprehensive characterization of the signaling dynamics of the RIG-I and interferon pathway to date (Burkart 2023). In this project, we have also approached the qualitative nature of viral antagonism (dampening vs. delaying the response) and learned a lot that turned out to be important also for countering SARS-CoV-2 (see below). Together with the group of Ursula Klingmüller here at DKFZ, we have previously also studied the dynamics and complex regulatory events in the downstream interferon signaling system (Robichon 2020; Kok 2020).
As for regulators of the cell-intrinsic antiviral system, we have performed a high-throughput screen for cellular kinases regulating the RIG-I signaling pathway several years ago. We described one kinase, DAKP1, to be an induced inhibitor of the sensor molecule RIG-I, contributing to the proper termination of the antiviral defense program (Willemsen 2017, Figure 1B). A follow-up investigating the effects of DAPK2 and -3 is currently in the making, and further >20 kinases were identified in this study and await in-depth characterization. Moreover, we were collaborating to study the role of ubiquitin-ligases TRIM25 and RIPLET in the activation of the RIG-I / IRF3 signaling pathway and found a surprisingly impactful role for RIPLET (Cadena 2019; Hayman 2019). In another siRNA screen, we furthermore identified several other ubi-ligases potentially playing a role in regulating the antiviral pathway, which are currently under scrutiny. At the level of transcription factors, we were curious about the roles of the different "interferon regulatory factors" (IRFs) in our beloved model cell line A549, beyond the well-established roles for IRF3 and IRF9 (Wüst 2021).
- Burkart SS, Schweinoch D, Frankish J, Sparn C, Wüst S, Urban C, Merlo M, Magalhães VG, Piras A, Pichlmair A, Willemsen J, Kaderali L, Binder M. (2023) High-resolution kinetic characterization of the RIG-I-signaling pathway and the antiviral response. Life Sci Alliance, 6(10):e202302059. doi: 10.26508/lsa.20230205
- Wüst S, Schad P, Burkart SS, Binder M (2021) Comparative Analysis of Six IRF Family Members in Alveolar Epithelial Cell-Intrinsic Antiviral Responses. Cells 10(10):2600
- Robichon K, Maiwald T, Schilling M, Schneider A, Willemsen J, Salopiata F, Teusel M, Kreutz C, Ehlting C, Huang J, Chakraborty S, Huang X, Damm G, Seehofer D, Lang PA, Bode JG, Binder M, Bartenschlager R, Timmer J, Klingmüller U (2020) Identification of Interleukin1β as an Amplifier of Interferon alpha-induced Antiviral Responses. PLoS Pathog, 16(10):e1008461
- Kok F, Rosenblatt M, Teusel M, Nizharadze T, Gonçalves Magalhães V, Dächert C, Maiwald T, Vlasov A, Wäsch M, Tyufekchieva S, Hoffmann K, Damm G, Seehofer D, Boettler T, Binder M, Timmer J, Schilling M, Klingmüller U (2020) Disentangling molecular mechanisms regulating sensitization of interferon alpha signal transduction. Mol Syst Biol, 16(7):e8955
- Cadena C, Ahmad S, Xavier A, Willemsen J, Park S, Park JW, Oh SW, Fujita T, Hou F, Binder M, Hur S (2019) Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 177(5):1187-1200.e16
- Hayman TJ, Hsu AC, Kolesnik TB, Dagley LF, Willemsen J, Tate MD, Baker PJ, Kershaw NJ, Kedzierski L, Webb AI, Wark PA, Kedzierska K, Masters SL, Belz GT, Binder M, Hansbro PM, Nicola NA, Nicholson SE (2019) RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol Cell Biol 97(9):840-852
- Willemsen, J., Wicht, O., Wolanski, J.C., Baur, N., Bastian, S., Haas, D.A., Matula, P., Knapp, B., Meyniel-Schicklin, L., Wang, C., Bartenschlager, R., Lohmann, V., Rohr, K., Erfle, H., Kaderali, L., Marcotrigiano, J., Pichlmair, A. and Binder, M. (2017) Phosphorylation-Dependent Feedback Inhibition of RIG-I by DAPK1 Identified by Kinome-wide siRNA Screening. Molecular Cell 65 (3):403415
II. Involvement of cell-intrinsic antiviral signaling in tumor development and therapy
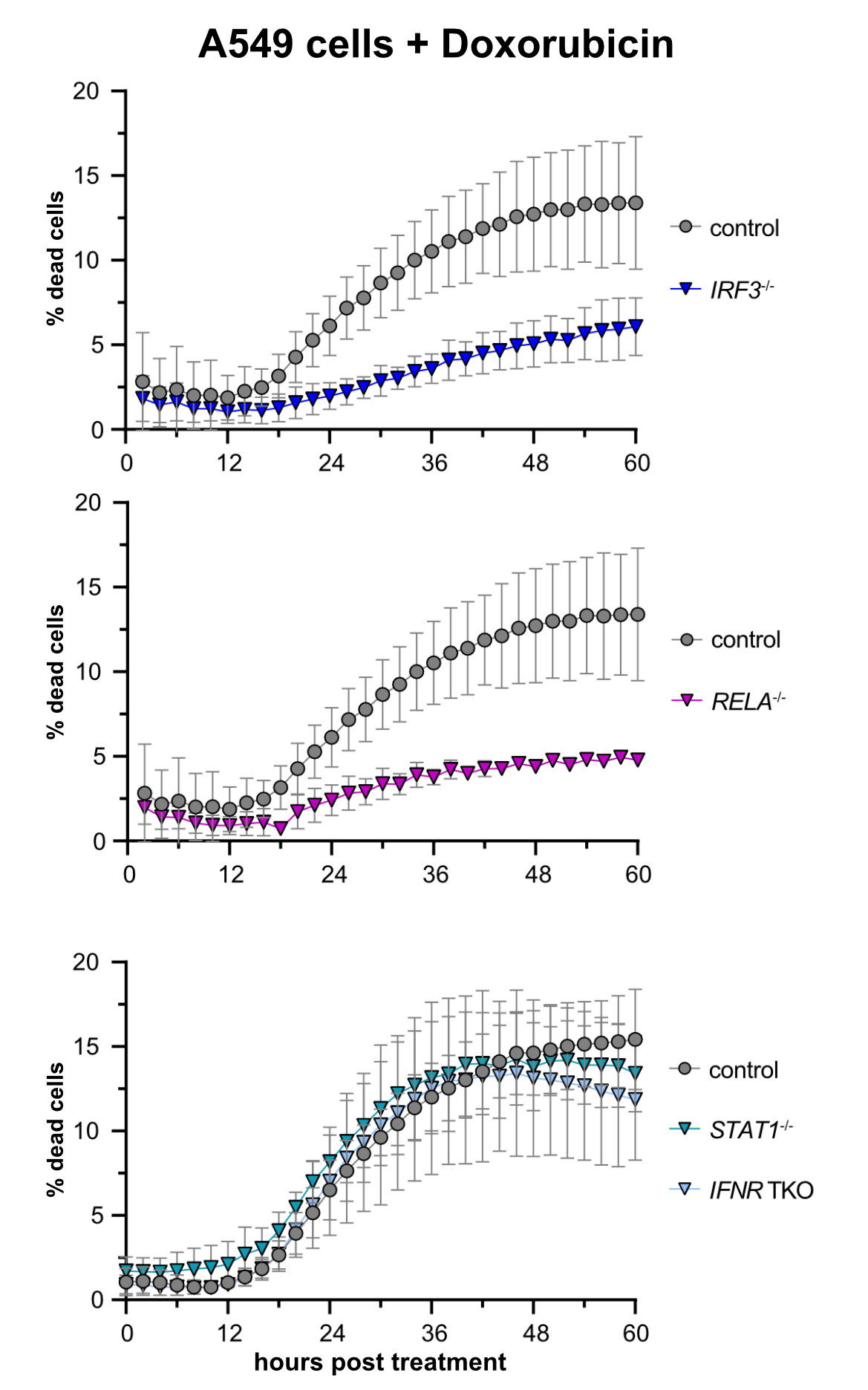
Treatment of A549 lung adenocarcinoma cells with doxorubicin (a chemotherapeutic) induces cell death. Remarkably, this cell death is largely dependent on the presence of intact RIG-I signaling, whereas signaling downstream of the interferon receptor appeared dispensable.
© dkfz.de
Cell-intrinsic antiviral immunity is pivotal to combat virus infections. Sensing of conserved pathogen-associated molecular patterns (PAMPS) by cellular receptors such as RIG-I results in the production and release of interferons and massive expression of interferon-stimulated genes (ISGs), conferring an antiviral state. Notably, this response not only comprises antiviral effectors but also displays cytostatic and anti-tumorigenic properties, inducing apoptosis, and suppressing proliferation and migration. Upon continued triggering of a very slight RIG-I signal, we observed significantly inhibited cell growth, which could be reversed by knockout of the transcription factor IRF3. Initial results indicated that this cytostatic effect was not (only) mediated by secreted interferons but rather was an intrinsic effect of the "infected" cell (Urban 2020). We are currently investigating whether prolonged stimulation of this antiviral system, as it occurs during chronic viral infection, would lead to selection and preferential outgrowth of interferon resistant cells. This could have significant implications for both viral persistence as well as the development of tumors in the long run. This project is part of a large DFG-funded "transregional" Collaborative Research Center, TRR179.
Recent literature has furthermore shown that antiviral innate immune signaling is directly involved in tumor development and/or immune control of emerging tumors, albeit the molecular mechanisms are not at all understood yet. Surprisingly, two studies (Sistigu et al., Nature Medicine 2014; Ranoa et al., Oncotarget 2016) have also demonstrated a direct involvement of the RIG-I/interferon axis in the induction of tumor-cell death in at least a subset of chemotherapies, e.g. doxorubicin. Based on these anecdotal reports, our research aims to identify and characterize the components and the mechanism of RIG-I mediated cell-death upon doxorubicin-induced DNA damage. We generated A549 CRISPR/Cas9 knockout cell lines targeting different components of innate immunity and determined rates of cell death upon treatment with cytostatics or also γ-irradiation by life cell imaging. We confirmed that doxorubicin treatment induces cell death dependent on a functioning RIG-I/MAVS axis, including the transcription factors IRF3 and NFkB, while the interferon system appeared dispensable (see figure), but a third antiviral transcription factor, IRF1, turned out to be surprisingly important (Zander 2022).
- Hiebinger F., Kudulyte A., Chi H, Burbano De Lara S, Ilic D, Helm B, Welsch H, Dao Thi VL, Klingmüller U, Binder M (2023, in press) Tumour cells can escape antiproliferative pressure by interferon-β through immunoediting of interferon receptor expression. Cancer Cell International doi:10.1186/512935-023-03150-y
- Pecori R, Ren W, Pirmoradian M, Wang X, Liu D, Berglund M, Li W, Tasakis RN, Di Giorgio S, Ye X, Li X, Arnold A, Wüst S, Schneider M, Selvasaravanan KD, Fuell Y, Stafforst T, Amini RM, Sonnevi K, Enblad G, Sander B, Wahlin BE, Wu K, Zhang H, Helm D, Binder M, Papavasiliou FN, Pan-Hammarström Q. (2023) ADAR1-mediated RNA editing promotes B cell lymphomagenesis. iScience 26(6):106864
- Zander DY, Burkart S, Wüst S, Gonçalves Magalhães V, Binder M. (2022) Cooperative Effects of RIG-I-like Receptor Signaling and IRF1 on DNA Damage-Induced Cell Death. Cell Death Dis. 13, 364
- Urban C, Welsch H, Heine K, Wüst S, Haas DA, Dächert C, Pandey A, Pichlmair A, Binder M. (2020) Persistent Innate Immune Stimulation Results in IRF3-Mediated but Caspase-Independent Cytostasis. Viruses 12(6):635
III. Understanding the dynamics of viral replication and its interplay with the cellular antiviral system
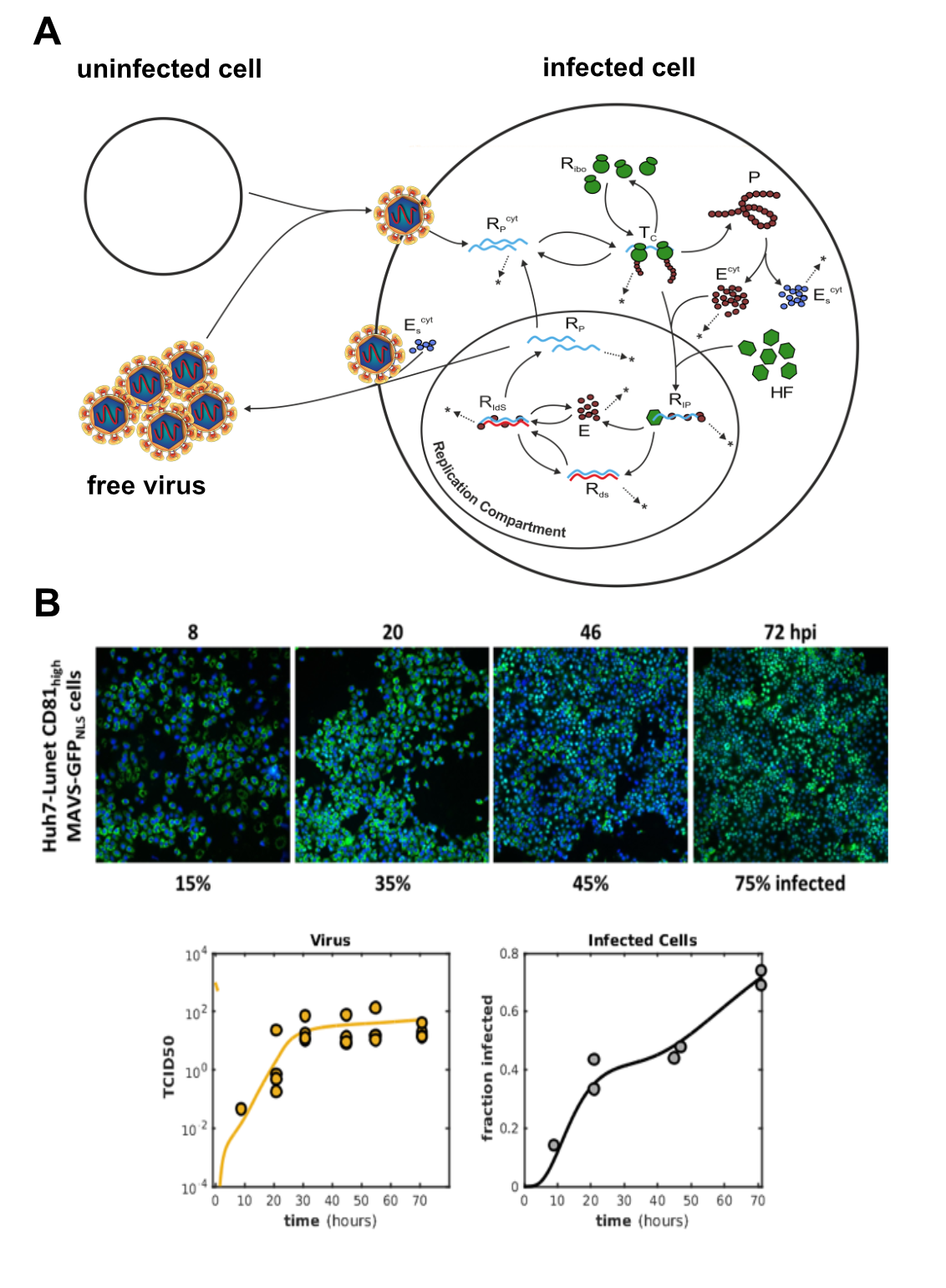
(A) Schematic representation of the extended full-lifecycle model of HCV replication. The intracellular core part of the model is based on our previously published work (Binder 2013). (B) The model was trained on experimental data acquired from authentic HCV infection experiments in our BSL-3 lab. We measured positive and negative strand viral RNA, viral protein production, secretion of offspring viral particles and monitored the number of productively infected cells over time. Circles in the plots represent actual measurements and the lines are model predictions.
© dkfz.de
The interplay between virus infection, establishment of replication and the induction of the cell-intrinsic antiviral defense program is at the core of the decision of whether a virus will be efficiently cleared or leads to productive infection and disease. The dynamics, i.e. promptness and vigor, of the two processes is critical if the cell-intrinsic response is quick enough, it can kill off the virus before it enters its exponential replication phase. In the contrary, as most (if not all) viruses encode factors that potently antagonize the cellular defense program, once the virus is one step ahead, it may effectively block the induction of the RIG-I/interferon response and manage to establish robust replication within the host cell. In order to understand this complex interplay, we teamed up with biomathematicians (Prof. Lars Kaderali, Greifswald) who together with us establish mathematical models (based on systems of ordinary differential equations) accurately simulating molecular pathways or virus replication cycles.
Several years ago, we have established the most detailed model of intracellular replication of the Hepatitis C virus (HCV) (Binder 2013). This model comprises every single major step of genome replication, beginning with translation of viral proteins, setting up replication organelles and replicating the viral RNA genome. Recently, we have been extending this original model by the extracellular steps of the HCV life cycle, i.e. the production and secretion of infectious viral particles from the host cell, and the infection of new "naive" host cells (figure 3A). The mathematical model is trained and validated against ample sets of experimental data. Generating this data required tedious measurements of viral parameters in our BSL-3 laboratory. The model can very precisely predict the course of amplification of a given amount of viral inoculum (figure 3B), and can be used to predict and understand the effects of various antiviral substances. In the future, we propose such modeling approaches to be used for establishing optimal drug regimens, meaning combinations of optimal doses of differently acting antiviral compounds.
We are going to set up models for further viruses; a model for Dengue virus has already been set up in collaboration with the team of Ralf Bartenschlager (Zitzmann 2020), and a more general model for the replication of plus-strand RNA viruses has currently been submitted (Zitzmann bioRxiv 2022). In parallel, we are setting up mathematical models of cellular antiviral pathways, most importantly the RIG-I signaling pathway. Based on previous biochemical studies of the pathway in our lab, we have recently published a model of RIG-I activation (Schweinoch 2020). Currently, we are finalizing a very large combined model of the RIG-I and the interferon JAK/STAT signaling cascades, allowing for the simulation of the full antiviral response over the course of 24 hours post infection (Burkart bioRxiv 2022, see above).
Our longer-term goal is to combine the models of viral replication with models of cell-intrinsic antiviral responses. This will eventually permit studying the mutual interactions as described above and potentially allow for a better understanding of the essential parameters determining the outcome of infection. A particular focus will be the difference between acute, self-limiting infections and infections that establish persistence, such as HCV, where a delicate balance of viral and antiviral processes must exist. This line of research is currently paused, but we nevertheless plan to work on it in the future! Please note that our lab is almost exclusively doing experimental work, with the modelling being done by the group of Lars Kaderali!
- Zitzmann C, Dächert C, Schmid B, van der Schaar H, van Hemert M, Perelson AS, van Kuppeveld FJM, Bartenschlager R, Binder M, Kaderali L (2023) Mathematical modeling of plus-strand RNA virus replication to identify broad-spectrum antiviral treatment strategies. PLoS Computational Biology 19(4):e1010423
- Schweinoch D, Bachmann P, Clausznitzer D, Binder M, Kaderali L (2020) Mechanistic modeling explains the dsRNA length-dependent activation of the RIG-I mediated immune response. J Theor Biol 500:110336
- Zitzmann C, Schmid B, Ruggieri A, Perelson AS, Binder M, Bartenschlager R, Kaderali L. (2020) A Coupled Mathematical Model of the Intracellular Replication of Dengue Virus and the Host Cell Immune Response to Infection. Front Microbiol. 11:725
- Binder, M., Sulaimanov, N., Clausznitzer, D., Schulze, M., Hüber, C. M., Lenz, S. M., Schlöder, J. P., Trippler, M., Bartenschlager, R., Lohman, V. and Kaderali L. (2013). Replication vesicles are load- and choke-points in the hepatitis C virus lifecycle. PLoS Pathog 9, e1003561
IV. Our Pandemic Special Investigations into the innate immune response against SARS-CoV2
Having worked on RNA viruses and their interaction with the host cell-intrinsic immune system for many years, in the light of the rampant pandemic of the newly emerged SARS-Coronavirus-2, we started a fourth research line in 2020. We have devoted one of our two BSL-3 labs to research on SARS-CoV-2 and began to study the interplay of this large RNA virus with the host cellular defense machinery. In a close collaboration with the team of Ralf Bartenschlager at the CIID Heidelberg, we were able to confirm that the virus very efficiently blocks the activation of the transcription factor IRF3 (see above), while still permitting activation of NFkB. This leads to a strongly biased transcriptional response, largely comprised of pro-inflammatory (but not antiviral) cytokines. This strong activation of pro-inflammtory factors was shown to be mediated through STING, an adapter molecule of the defense system usually triggered by DNA viruses (Neufeldt 2022).
In a second collaboration, we contributed to a large single cell RNA-sequencing study performed at the Charité hospital in Berlin. They examined the effects of hypertension (HT) and other cardiovascular disease (CVD) in the clinical course of COVID-19. It was known that HT/CVD significantly worsens the outcome of disease, but interstingly, we found that treatment with anti-hypertensive drugs ameliorated these detrimental effects. In particular inhibitors of the angiotensin converting enzyme I (ACEi) almost completely reverted the increased risk, leading to viral clearance comparable to non hypertensive patients. We found that secretory cells (the target cells for SARS-CoV2) in ACEi treated patients show a clear signature of cell-intrinsic antiviral signaling, indicating that this response might contribute to the positive outcome in this cohort (Trump 2020). In a second study of our colleagues at Charité, we identified the cell-intrinsic antiviral system to be much stronger and pre-activated in the nasal airway mucosa of children as compared to adults. Upon infection with SARS-CoV-2, this translates into a faster and much stronger inetrferon response in children, very likely being a major reason for their much higher resiliance to developing severe COVID-19 (Loske 2022).
In our latest study, we went into the molecular detail of how cells recognize SARS-CoV-2 and how for this virus the race between the kinetics of the cellular defense and viral replication and expression of antagonists absolutely critically determined the outcome of infection. We found an exciting interplay of immune cells in the nasal mucosa with epithelial cells, leading to a substantial speed-up of the antiviral responsiveness of epithelial cells (the targets of SARS-CoV-2 infection).
- Magalhães VG, Lukassen S, Drechsler M, Loske J, Burkart SS, Wüst S, Jacobsen EM, Röhmel J, Mall MA, Debatin KM, Eils R, Autenrieth S, Janda A, Lehmann I, Binder M (2023) Immune-epithelial cell cross-talk enhances antiviral responsiveness to SARS-CoV-2 in children. EMBO Reports e57912
- Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee J, Plociennikowska A, Heigwer F, Joecks S, Burkart SS, Zander DY, Gendarme M, El Debs B, Halama N, Merle U, Boutros M, Binder M, Bartenschlager R (2022) SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Commun Biol 5(1):45
- Loske J, Röhmel J, Lukassen S, Stricker S, Magalhaes VG, Liebig J, Chua RL, Thürmann L, Messingschlager M, Seegebarth A, Timmermann B, Klages S, Ralser M, Sawitzki B, Sander LE, Corman VM, Conrad C, Laudi S, Binder M*, Trump S*, Eils R*, Mall MA*, Lehmann I* (2022) Pre-activated antiviral innate immunity in the upper airways controls early SARS-CoV-2 infection in children. Nature Biotech 40(3):319
- Trump S*, Lukassen S*, Anker MS*, Chua RL*, Liebig J*, Thürmann L*, Corman VM*, Binder M*, Loske J, Klasa C, Krieger TG, Hennig BP, Messingschlager M, Pott F, Kazmierski J, Twardziok S, Albrecht JP, Eils J, Hadzibegovic S, [...] Lehmann I (2020) Hypertension delays viral clearance and exacerbates airway hyperinflammation in patients with COVID-19. Nature Biotech 39(6):705