Functional
Genome Analysis (B070)
Deutsches
Krebsforschungszentrum,
Im Neuenheimer Feld 580
D-69120
Heidelberg,
Germany. |
 |
.
.
|
Archive |
Transcriptional
Profiling Analyses |
|
 |
|
Melanoma
microRNA trafficking controls tumour primary niche formation



r
Melanoma
originates in the epidermis and becomes
metastatic after invasion into the dermis. Prior interactions between
melanoma
cells and dermis are poorly studied. Here, we show that melanoma cells
directly
affect the formation of the dermal tumour niche by microRNA trafficking
before
invasion. Melanocytes, cells of melanoma origin, are specialized in
releasing
pigment vesicles, termed melanosomes. In melanoma in situ, we found melanosome
markers in distal fibroblasts before melanoma invasion. The melanosomes
carry
microRNAs into primary fibroblasts triggering changes, including
increased
proliferation, migration and pro-inflammatory gene expression, all
known
features of cancer-associated fibroblasts (CAFs). Specifically,
melanosomal
microRNA-211 directly targets IGF2R and leads to MAPK signalling
activation,
which reciprocally encourages melanoma growth. Melanosome release
inhibitor
prevented CAF formation. Since the first interaction of melanoma cells
with
blood vessels occurs in the dermis, our data suggest an opportunity to
block
melanoma invasion by preventing the formation of the dermal tumour
niche.
.
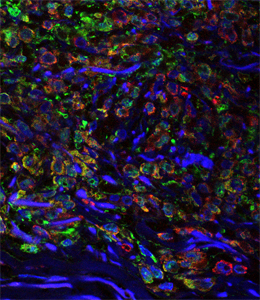
Figure legend.
Melanoma tumour in situ. The
melanoma cells have not yet left the
epidermis; green: fibroplasts; red: melanosomes; blue: DNA in cell
nuclei.
rr
rr
Dror, Sanders et al. (2016) Nature Cell Biol., in press. (doi:
10.1038/ncb3399). |
..
..
..
| Early
epigenetic down-regulation of microRNA-192 expression promotes
pancreatic cancer progression |
 |
r
Pancreatic
ductal adenocarcinoma (PDAC) is
characterized by very early metastasis, suggesting the hypothesis that
metastasis-associated changes may occur prior to actual tumor
formation. We
identified miR-192 as an epigenetically regulated suppressor gene with
predictive value in this disease. miR-192 was downregulated by promoter
methylation in both PDAC and chronic pancreatitis (CP), the latter of
which is
a major risk factor for development of PDAC. Functional studies in vitro and in vivo in mouse models of
PDAC showed that overexpression of
miR-192 was sufficient to reduce cell proliferation and invasion.
Mechanistic
analyses correlated changes in miR-192 promoter methylation and
expression with
epithelial-mesenchymal transition (EMT). Cell proliferation and
invasion were
linked to altered expression of the miR-192 target gene SERPINE1
that is encoding the protein plasminogen activator
inhibitor-1 (PAI-1), an established regulator
of these properties in PDAC cells. Notably, our data suggested that
invasive
capacity was altered even before neoplastic transformation occurred, as
triggered by miR-192 downregulation. Overall, our results highlighted a
role for
miR-192 in explaining the early metastatic behavior of PDAC and
suggested its
relevance as a target to develop for early diagnostics and therapy.
.
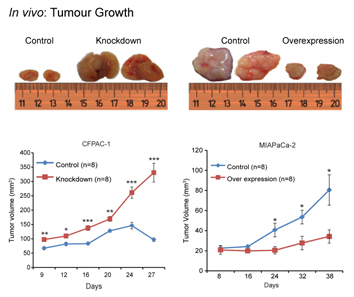
Figure legend.
Two cell lines, which express miR-192 at high level (CFPAC-1) or low
level (MIAPaCa-2) were transfected with constructs that suppressed or
increased miR-192 expression, respectively. Xenografted into mice, strong effects on
tumour growth were observed.
rr
Botla et al. (2016) Cancer Res. 76, 4149-4159.  |
|
 |
..
| Blood-borne
microRNA
signatures in human pathologies |
|
r
Beyond
studies that describe microRNAs frequently as
markers for specific traits, we asked whether a general pattern for
microRNAs
across many diseases exists that could act as an intial, non-invasive means of
diagnostics. In a
multicenter study, we evaluated
circulating
profiles of
1,049 patients suffering from 19 different cancer and non-cancer
diseases as
well as unaffected controls. The results were validated on 319
individuals from
three different centers using qRT-PCR. We detected consistently deregulated
profiles for
particular diseases; pathway analysis confirmed disease association of
the
respective microRNAs. Overall,
we discovered 34 miRNAs with
strong
disease association. In addition, a set of microRNAs was
discovered that act as rather stable
markers, offering
reasonable control microRNAs for
future studies. Our study further
underscores the
high potential of specific blood-borne miRNA patterns as molecular
biomarkers.
.
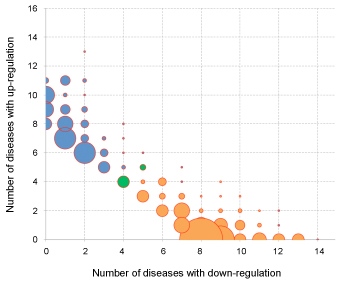
Figure legend.
The
balloon plot shows in how
many diseases microRNAs are up- or down-regulated. The bubble size
represents the
number of microRNAs showing the respective distribution of up- and
down-regulation. Orange bubbles represent microRNAs that are
predominantly
down-regulated, while blue bubbles stand for predominantly up-regulated
microRNAs.
The two green bubbles represent 9 microRNAs that were equally often up-
and down-regulated in disease.
rr
Keller et al. (2014) BMC Med. 12,
224.
Keller et al. (2011) Nature
Meth. 8,
841-843.
|
|
|
r
rr
MicroRNAs in the
blood of patients with primary CNS lymphoma (PCNSL) act as prognostic
biomarkers
rr
Despite
improved therapeutic regimens, primary CNS lymphoma (PCNSL) remains a
therapeutic challenge. We
characterized by next generation sequencing the microRNA fingerprints
in the
blood of PCNSL patients enrolled in a large randomized phase III study,
comparing
short-term to long-term survival. Twelve microRNAs were significantly
deregulated between the two groups. The microRNAs miR-493-3p and
miR-432-5p
exhibited the most prominent differences. Furthermore, we identified
short RNA
molecules with putative microRNA function that had not been described
before.
.
Roth et al. (2015) Eur. J. Cancer 51, 382-390.
|
|
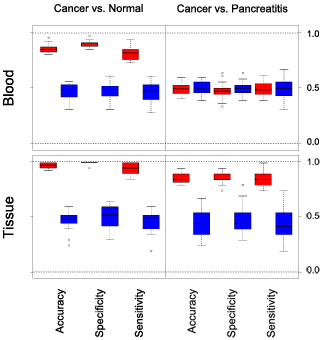
Boxplot presentation of the pancreas classification results
|
|
| Diagnosis
of pancreatic ductal adenocarcinoma and chronic pancreatitis by
measurement of microRNA abundance in blood and tissue |
|
r
MicroRNAs
can regulate hundreds of genes post-transcriptionally and appear to
regulate virtually all cellular processes. Owing to these properties,
microRNAs
have a critical role not only in physiological but also in
pathological processes.
There is meanwhile a lot of evidence that miRNA profiles from body
fluids, such as blood, and informative about the disease status of a
blood donor.
r
A detailed analysis of microRNA profiles was performed
for
pancreatic ductal adenocarcinoma. A solid diagnostic process could have
substantial impact on the successful treatment of the disease, for
which
currently mortality is nearly identical to incidence. Variations
in the abundance of all microRNA
molecules from peripheral blood cells and pancreas tissues were
analysed. In total, 245
samples from two clinical centers were studied that were obtained from
patients
with pancreatic ductal adenocarcinoma or chronic pancreatitis and from
healthy
donors.
r
Thee blood test demonstrated very
high sensitivity and specificity of a distinction between healthy
people and
patients with either cancer or chronic pancreatitis. Confirmative and
partly even more
discriminative diagnosis could be
performed on tissue samples. In
addition, discrimination between cancer and chronic pancreatitis was
achieved. Several miRNAs were identified that exhibited
abundance variations in both tissue and blood samples.
r
The results
could have
an immediate diagnostic value for the evaluation of tumour reoccurrence
in
patients, who have undergone curative surgical resection, and for
people with a
familial risk of pancreatic cancer.
r
Bauer et al. (2012) PLoS ONE 7,
e34151.
|
|
| MicroRNA
signatures
of peripheral blood cells in humans infected with Trypanosoma brucei gambiense |
 |
r
Human
African Trypanosomiasis -
sleeping sickness - still affects thousands of people a year. Control
relies on
diagnosis and treatment, and in the absence of this surveillance, the
number of
cases rapidly rebounds. Diagnosis relies on an antibody test and
microscopy.
The type of treatment depends on the disease stage: whether parasites
have
entered the central nervous system; and this can be determined only by
lumber
puncture and analysis of cerebrospinal fluid. The development of
sensitive,
simple and reliable tools for diagnosis and staging of human African
Trypanosomiases would significantly ease field surveillance and enhance
patient
care. We investigated whether the patterns of miRNAs from peripheral
blood
could be used to decide whether patients are infected, or to determine
the
disease stage. We found that the miRNA patterns did differ between
parasite-positive patients and uninfected controls with no immune
reaction to
trypanosomes. Also, people with immune reactions to trypanosomes, but
no
detectable parasites, sometimes showed patient-like profiles. The
patterns were
not reliable enough, however, to be used for diagnosis.
rr
Lueong et al. (2013) PLOS ONE 8,
e74555.
|
a |
|
MicroRNA
expression profiles in peripheral blood cells of rats infected with Trypanosoma congolese and Trypanosoma brucei subspecies
r
To identify
differentially regulated miRNAs during
trypanosome infections, we analyzed the miRNA
expression profiles of uninfected rats and animals infected with Trypanosoma congolense and different Trypanosoma
brucei species. The
potential target
genes of the regulated miRNAs as well as their biological pathways and
biological functions were investigated.
Simo et al. (2015) Microb. Infect. 17, 596-608.
|
|
 |
|
..
MicroRNA
variations in cells and their exosomes upon chemotherapy
r
It has been convincingly proposed that exchange of
molecules via exosomes is a means of eukaryotic intercellular
communication,
especially within the tumour microenvironment. However, there are no
data on
the alterations of exosomal molecular cargo upon pharmacological
anticancer
treatments. To approach this issue, we analysed the abundance of miRNAs
and
cancer-related proteins in exosomes secreted by Caco-2
(Cetuximab-responsive)
and HCT-116 (Cetuximab-resistant) colorectal cancer cells before and
after Cetuximab
treatment. We also characterized both profiles in whole source cells.
r
Cetuximab significantly altered the molecular cargo
of exosomes from Caco-2: we detected increased abundance of miRNAs and
proteins
that activate cell proliferation and proinflammatory processes;
simultaneously,
we observed a decrease of miRNAs and proteins related to immune
suppression.
These changes did not overlap entirely with those in source cells,
suggesting a
Cetuximab-linked distribution bias.
r
Molecular changes of a minor extent were also
detected in exosomes from HCT-116. Transfection of exosomes from
Cetuximab-treated Caco-2 into HCT-116 significantly increased HCT-116
viability; conversely, Caco-2 transfected with exosomes from treated
HCT-116
did not show viability alterations. This suggests that the molecular
phenotype
of source cells is important for determining both the exosomal cargo as
the
biological effects of transferred exosomes.
r
Gene Ontology analysis of networks that comprise
targets of differentially expressed exosomal miRNAs and proteins
demonstrated a
significant involvement of biological processes related to
proliferation
control, inflammation, immune response, and apoptosis. Our data shed
light on
molecular mechanisms of intercellular communication in eukaryotes.
Ragusa et al. (2014) Oncoscience 1,
132-157.
|
|
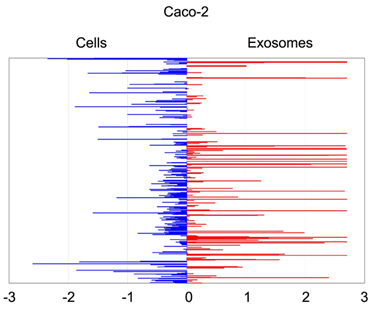
Quantitative
asymmetric
distribution of miRNAs in colorectal cancer cells and exosomes.
Relative
quantities (RQ) of the miRNAs amounts as isolated from exosomes were
compared to those from the
source cells Caco-2. Values are shown as log10 of RQ.
|
FINISHED PROJECT:
Identification
of
malignancy factors by analysing cystic tumours of the pancreas
|
|
  
|
|
|
|
The
diversity in the
aggressiveness of cystic tumours of the pancreas – ranging from the
usually
benign serous cystadenoma to lesions of variable degrees of malignancy
– was
utilised for the identification of molecular factors that are involved
in the
occurrence of malignancy. We analysed the transcript profiles of
different
cystic tumour types. Variations could be identified that could be
critical for
the regulation of malignancy and thus relevant to the treatment of also
the
majority of pancreatic tumours. The results
were confirmed at the protein level by immunohistochemistry.
Also, functional studies with siRNA silencing were performed.
Expression variations at the RNA and
protein level were identified that are closely correlated with the
degree of
malignancy. Besides, all tumours could be classified effectively by
this means.
Many of the identified factors had not previously been known to be
associated
with malignant cystic lesions. SiRNA silencing of the gene with the
most
prominent variation – the anti-apoptotic factor FASTK (Fas-activated
Serine/Threonine Kinase) – revealed a regulative effect on several
genes known
to be relevant to the development of tumours.
Bauer et al.
(2009) Pancreatology 9, 34-44.
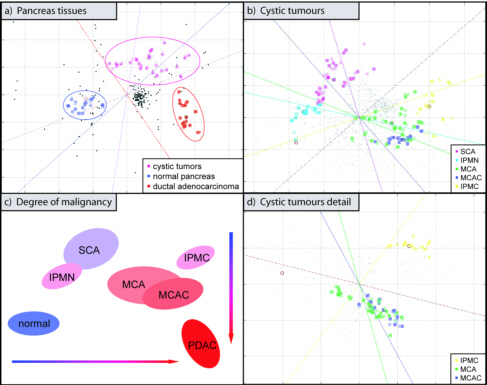
Figure legend: correspondence
cluster
analysis of transcript profiles. In the resulting biplot, each
hybridisation of
an individual sample is depicted as a coloured square. Genes that
exhibited
significantly differential transcription levels are shown as black
dots. The
closer the co-localisation of two spots (both genes and tumours) the
higher is
the degree of association between them. Also, guidelines are displayed
in the
diagram. They are calculated from the data and point to the positions
of
virtual genes, which exhibit a variation in one tumour entity only. The
closer
a depicted gene lies to one of these guidelines and the further its
distance to
the centroid the better its expression is described by the respective
ideal profile.
All genes that are not significantly differentially transcribed are
located
close to the centroid of the lines but are not shown for clarity. In (a), a cluster analysis is shown of
normal pancreatic tissue, all cystic tumours combined and ductal
adenocarcinoma. Panel (b) presents
the results obtained for the cystic tumours alone. As a consequence of
the
normalisation process, only the median of the controls is shown in the
diagram
as a single red circle instead of the individual hybridisation events.
In (d), a close-up of the data is shown,
which were generated with the IPMC, MCA and MCAC samples. Panel (c), finally, presents a combination of
the data with a colour-code added that indicated the tendency of
malignancy of
the respective tumour types: blue, non-malignant; red, highly malignant.
|
|
 |
 . .
Collaborators:
Frank Sauer &
Renato Paro

|
|
FINISHED PROJECT:
Functional
analysis
of gene and protein networks in Drosophila with microarray
based
expression studies
The complete
genomic sequence of several metazoan
organisms such as Drosophila melanogaster and Caenorhabditis
elegans
was available. The next task arising from this sequence data was (and
still is) the
deciphering
of the role and function of the identified genes and their
corresponding
protein products in the context of an entire organism. As often the
expression
pattern of a gene provides clues to its function, we produced a
DNA-microarray
that enabled us to monitor gene expression in the context of the entire
Drosophila
genome. The system enabled us to identify genes whose
activities are
required for the execution of complex developmental gene networks and
signal
transduction pathways. As such networks and pathways are evolutionary
highly
conserved among metazoans, the analyses of gene and protein function in
Drosophila
also provided valuable clues for a better knowledge of
corresponding
pathways in vertebrates.
While
the genome sequences for a variety of
organisms are now available, the precise number of genes encoded is
sometimes still a
matter of debate. We based our whole-transcriptome microarray, the
Heidelberg
FlyArray, on the combination of the BDGP annotation and a novel ab initio gene prediction of lower
stringency using the Fgenesh software. A
microarray was established with altogether some 24,000 different
features, each
actually present in duplicate. The primer
set used to produce the PCR-products was from Eurogentec.
Apart from being used
for the production
of the microarray, the very primer set was also applied to the
generation of a genome-wide dsRNA library, the actual work being
performed in the laboratory of Norbert Perrimon
at Harvard Medical School in Boston (USA). This molecule set
allowed the
identification of gene functions by cell-based RNAi-screens.
|
|
To
assess
the overall quality of our array design
as well as to validate the novel predictions, we performed
developmental
profiling of the Drosophila lifecycle
using 9 different stages. We
were able to provide evidence for the transcription of ~2,600
additional genes
predicted by Fgenesh. Validation of the developmental profiling data by
RT-PCR
and in situ hybridization indicated a
lower limit of 2,000 novel annotations, thus substantially raising the
number of
genes that make a fly. The successful design and application of this Drosophila microarray confirmed our
expectation that mere in silico
approaches will always tend to be incomplete. The identification of at
least
2,000 novel genes highlights the importance of gathering experimental
evidence
to discover all genes within a genome. Moreover, as such an approach is
independent of homology criteria, it will allow the discovery of novel
genes
unrelated to known protein families or which have not been strictly
conserved
between species.
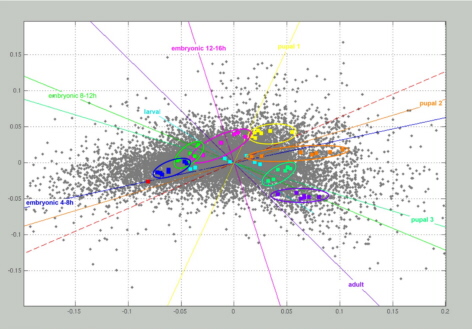
Figure to the right.
Correspondence
cluster analysis of the
developmental profiling. Samples from 9 different stages of the Drosophila lifecycle were hybridised. Each hybridisation
of an individual developmental stage is depicted as coloured
square. They all form distinct clusters
– but for the larval stage – indicating the degree of
reproducibility and
specificity between them. As a consequence of the normalisation
process, only
the median of all control hybridisations (0-4 h) is shown in the
diagram as a
single red square. Genes are shown as grey dots, if they exhibited
significant
differential transcription levels. The distance between dots is low
when their
expression profiles show similar shape, independent of their absolute
values.
|
|
|
Diehl et
al. |
(2002) |
Nucleic
Acids Res. 30, e79. |
 |
|
|
Hild et
al. |
(2003) |
Genome
Biol. 5, R3. |
|
|
|
Boutros et al. |
(2004) |
Science 303, 382-385. |
|
|
|
Altenhein et al.
|
(2006)
|
Develop.
Biol. 296, 545-560.
|
|
|
|
|
|
|
 |
FINISHED PROJECT:
Creation of
a minimal tiling
path of genomic clones for Drosophila melanogaster;
provision
of a common resource



On the
basis of shotgun subclone libraries used in the sequencing of the Drosophila melanogaster genome, a
minimal tiling path of subclones across much of the genome was
determined.
About 320,000 shotgun clones for chromosomes X(12-20), 2R, 2L, 3R and 4
were
available from the Berkeley Drosophila
Genome Project. The clone inserts have an average length
of 3.4 kb and are amenable to standard PCR-amplification. The resulting
tiling
path covers 86.2% of chromosome X(12-20), 86.2% of chromosomal arm 2R,
79.0% of
2L, 89.6% of 3R and 80.5% of chromosome 4. In total, the 25,135 clones
represent 76.7 Mb – equivalent to about 67% of the genome – and would
be
suitable for producing a microarray on a single slide.
This work was performed in collaboration with Susan
Celniker (Berkeley), Eric
Johnson
(University
of Oregon) and Eileen
Furlong (EMBL).
Hollich
et al. (2004) Biotechniques 37, 282-284.
|
|
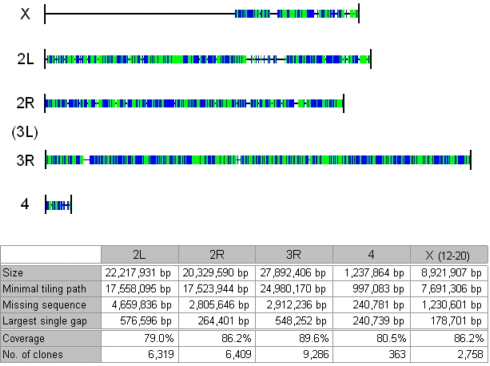
Schematic
representation of the tiling path’s genome coverage. Horizontal lines
indicate
the chromosomes of the 115 Mb Drosophila
genome. The genomic regions that are covered by the minimal tiling path
of
25,135 shotgun clones are represented as blue and green coloured areas.
Interruption of the colouring depicts large gaps. Any change in colour
from blue
to green or visa versa indicates the
existence of a gap that is too small to be visible. Below, a table
presents the
relevant numbers.
|
...
|
|
|
|
|
Hauser et
al. |
(1998) |
Yeast
14, 1209-1221. |
|
|
Hauser et
al. |
(2002) |
Screening 3/02, 28-31. |
|
|
|
|
|
Hauser et
al. |
(1998) |
Meth.
Microbiol.28, 193-204. |
|
|
Yin et
al. |
(2003) |
Mol.
Microbiol. 48, 713-724. |
|
|
|
|
|
Beissbarth et
al. |
(2000) |
Bioinformatics 16,
1014-1022. |
|
|
Lagorce et
al. |
(2003) |
J.
Biol. Chem. 278, 20345-20357. |
|
|
|
|
|
Hoheisel
& Vingron |
(2000) |
Res.
Microbiol. 151, 113 -119. |
|
|
Fellenberg et
al. |
(2003) |
Perspect. Gene Expression,
Eaton Publ., 307-343. |
|
|
|
|
|
Fellenberg et
al. |
(2001) |
Proc.
Natl. Acad. Sci. USA 98, 10781-10786. |
|
|
Becerra et
al. |
(2003) |
Comp.
Funct. Genomics 4, 366-375. |
|
|
|
|
|
Hauser et
al. |
(2001) |
Comp.
Funct. Genomics 2, 69-79. |
|
|
Hagen et al. |
(2004) |
Mol.
Microbiol. 52,
1413 -1425. |
|
|
|
|
|
Fellenberg et
al. |
(2002) |
Bioinformatics 18,
423-433. |
|
|
Busold et al. |
(2005) |
Bioinformatics 21, 2424-2429. |
|
|
|
|
|
Becerra et
al. |
(2002) |
Mol.
Microbiol. 43, 545-555. |
|
|
Fellenberg et
al. |
(2006) |
BMC
Genomics 7, 319. |
|
|
|
|
|
Lombardia et
al |
(2002) |
Cell
Calcium 32, 83-91. |
|
|
Hauser et
al. |
(2007) |
FEMS Yeast
Res. 7, 84-92. |
|
|
|
|
|
|
FINISHED
PROJECT:
"MouseExpress":
In silico
analysis of expression profiles in mouse-mutants
.
..
Collaborators:
Johannes
Beckers , Martin
Hrabe de Angelis
, GSF, Munich; Werner
Mewes , GSF, Munich; Martin
Vingron , MPIMG, Berlin.
|
The sequencing of
the mouse
and human
genomes have basically been accomplished. The next step for the
integration of
this genomic information into biological and biomedical research will
be the systematic analysis of gene function. The similarities between
man
and mouse in their genomes, molecular pathways, physiology and
developmental
mechanisms make the mouse the most important model organism for the
study
of inherited diseases in man.
...
To
discover new mutants that
serve as
models for
human diseases or that have developmental defects, mice that have been
subjected to ENU mutagenesis are routinely examined for clinical
parameters,
behaviour and dysmorphologies (Mouse ENU Mutagenesis Screen, Institute
of Experimental Genetics, GSF, Neuherberg-Munich, Germany). In the
'MouseExpress'
project, we extended phenotypic description to a molecular level.
Combining DNA microarrays and appropriate phenotypical data, we
compared RNA
expression
profiles of thousands of genes from tissues or embryos of mutant and
wildtype
mice.
...
We used the RNA
expression profiles to identify molecular pathways
that are affected in disease and analysed interdependencies between
pathways
within a molecular network. Expression profiles of mutant and wildtype
mouse strains are filed in a database and will be linked to the
phenotype
and mutant databases of the Mouse ENU Mutagenesis Screen of the GSF.
|
|
 |
Beckers et al. (2002) Curr.
Genet. 3, 121-129. .
|
FINISHED PROJECT:
Transcriptional
profiling of Arabidopsis thaliana
 .
.
Global transcriptional
profiling in Arabidopsis
thaliana was
started in the EU-funded PPMdb-network and extended as part of the
German
ZIGIA consortium. Analyses were initially performed on a set of some
13,000
non-redundant EST-clones combined from the EST-clone collection of the
Institut National de la Recherche Agronomique (Versailles, France) and
the MSU EST-clone collection
obtained
from the Arabidopsis Biological Resource Center at the Ohio State
University
(Columbus, USA). Subsequently, analyses were performed on a set of
50mer
oligonucleotides, representing the Arabidopsis
gene set.
Various conditions were
studied. One initial area of emphasis was
the analysis of pathogen responses, done
in collaboration with Nikolaus
Schlaich and Alan
Slusarenko
of the RWTH Aachen.
Further studies were performed
in a local collaboration with Florian
Haas and Rüdiger Hell
of the Heidelberg Institute for
Plant Science (HIP) at Heidelberg University. They aimed at the
elucidation of the effects of mitochondrial serine acetyltransferase
functions on cysteine synthesis in plant cells. At the cellular level, cysteine synthesis in plants is entirely
different from that in non-photosynthetic eukaryotes.
Scheideler et al. (2002) J. Biol. Chem. 277, 10555-10561.
.
Wambutt et al. (2000) J. Biotechnol. 78,
281-292.
.
Haas et al. (2008) Plant
Physiol. 148, 1055-1067.
.
-------------------------------------------------------------------------------
other publications
and patents
|
|
 |
..
FINISHED PROJECT:
Analyses
in Trypanosoma brucei
|

|
|
|
|
|
|
The life
cycle of Trypanosoma brucei
involves
adaptation to a variety of conditions in the host and Tsetse fly. The
successive changes in morphology, biochemistry and plasma membrane
proteins, some
of which also involve cell cycle arrest, are still poorly
understood. Many
of these changes are likely to be directed by changes in mRNA abundance
and
translation, and over two decades of effort have now been expended in
the
identification of stage-specific mRNAs. Because of technical
limitations,
however, most of the transcripts identified have been rather abundant,
and the
regulation studied has mainly been restricted to the rapidly-dividing
long
slender bloodstream and procyclic forms.
...
Given the
relatively small size of the T.
brucei
genome, there is a good prospect of a complete exploration of its
genome by
microarray analyses. This will allow identification of lower-abundance
regulated transcripts, and (using amplification methods) the study of
the
transcriptome of the less accessible forms found in the Tsetse fly. One
format
for the analysis is to perform genome-wide expression studies on
genomic
instead of gene-specific fragments. Such arrays have some intrinsic
advantages. Overall genome representation is usually good in shotgun
libraries
with comparatively little variation across the genome. Thus, even if
randomly
selected clone inserts are used as probes, there should be good
coverage and
relatively little redundancy. Also, not only coding but also intergenic
regions
can be studied, for example in chromatin immunoprecipitation
experiments.
Insert amplification can be performed with a single primer pair and
functional
analyses can actually precede sequencing.
...
In
collaboration with the group of Christine
Clayton, we
produced DNA-microarrays containing more than
21,000 PCR-products of 2 to 2.5 kb long genomic fragments of T. brucei strain
TREU927/4, which were used in several projects. We
also applied oligonucleotide microarrays provided by TIGR. |
|
 |
| Diehl et al. |
(2002)
|
Mol. Biochem. Paras. 123, 115-123.
|
|
| Brems et
al. |
(2005) |
Mol. Biochem. Paras. 139, 163-172. |
|
| Van-Duc et
al.
|
(2006) |
Mol. Biochem. Paras. 150, 340-349. |
|
| Denninger et
al.
|
(2007) |
Exp. Cell.
Res. 313, 1805-1819. |
|
| Hartmann et
al. |
(2007) |
Eurcaryotic
Cell 6, 1964-1978. |
|
| Kramer et
al. |
(2008) |
J. Cell. Sci. 121, 3002-3014. |
|
|
| Clayton et
al. |
.(2008) |
Biochem. Soc.
Trans. 36, 520-521. |
|
| Wurst et
al. |
.(2009) |
Mol. Biochem. Paras. 163, 61-65. |
|
| Queiroz et
al.
|
.(2009) |
BMC Genomics 10,
495 |
|
| Archer et
al.
|
.(2009) |
PLoS Pathogen 5,
e1000565. |
|
| Kramer et
al. |
.(2010) |
J. Cell. Sci. 123,
699-711. |
|
|
FINISHED PROJECT:
Analysis of
gene
expression in Trypanosoma brucei
gambiense
Human
African Trypanosomiasis (HAT) is
a disease that exists in two forms. The acute form is caused by T. b. rhodesiense (observed in East Africa)
while the chronic form is due to T. b.
gambiense (found in West and central Africa).
Around 60 million persons are exposed to this disease with some 500,000
current
infections. Despite a relentless fight against HAT during the last
century, it
is currently resurgent in epidemic form in several countries.
The low
sensitivity of the diagnostic
techniques associated to the low parasitaemia that characterizes T. b. gambiense infections lead
permanently to residual human reservoir of trypanosomes in HAT foci
after
medical surveys. Moreover, the treatment of T.
b. gambiense infections requires toxic drugs that are also less
effective
in the late stage of the disease. This ineffective treatment is
strengthens by
the emergency of parasite strains resistant to the most available drug
(Melarsoprol). Since treatment of HAT is harsh and sometimes
ineffective, the
vaccine approach is probably a good perspective for trypanosomiasis
prevention.
Until now, there is no prospective vaccine for HAT despite the fact
that
investigations on vaccine have been a goal for nearly a century.
With the
resurgence of HAT, it is
important to improve the control of this disease by undertaking studies
that
may lead to the discovery of new genes essential for the survival of
trypanosomes. These genes could be targeted further for drugs, vaccine
or
diagnostic tools. The considerable differences between T.
b. gambiense and the others T.
brucei sub-species should be most likely the result of changes at
the nucleotide
sequences or in gene expression. We were analysing gene expression in T. b. gambiense using DNA-microarrays,
which were produced on the basis of the shotgun clones made for
sequencing. The
identification of sub-species specific gene expression variations could
greatly
facilitate the generation and testing of hypotheses on the mechanism of
human
serum resistance in T. b. gambiense,
the key factors to its ability to infect human.
Simo et al.
(2010) Infect. Genet. Evol. 10,
229-237.
|
FINISHED PROJECTS:
Transcription
analysis in microbial organisms
| Bacillus
subtilis |
 |
|
Within
a 'Leitmotiv Medizin' project, comparative studies were
performed in collaboration with Michael Hecker of
the University of Greifswald
on
the
variation of all
transcripts
of Bacillus subtilis -
carried out by microarray analysis - and the actual protein levels as
identified in 2D-electrophoresis.
...
Petersohn et al. (2001) J. Bacteriol. 183,
5617-5631.
.
| Neurospora
crassa |
|
|
Ever since
Tatum and Beadle formulated
their one-gene-one-enzyme hypothesis on the basis of studies with Neurospora crassa, this filamentous
fungus served as a model organism not only in genetics but also many
other
fields of basic research. Despite a lot of successful research, only
about one
tenth of the genes of Neurospora crassa
had
been described and localised on the seven chromosomes prior to genome
initiatives. Genome analysis started by ordering cosmid and BAC clones
along
individual chromosomes. Based on the physical clone maps
of linkage
groups II
and V, sequencing of the two chromosomes was done as part of the German
Neurospora Genome Project. Simultaneously, a
whole-genome
shotgun approach was taken at the Whitehead Genome Center, Cambridge, USA,
recently
yielding the complete genomic sequence.
...
For an
initial insight into
transcriptional variations in Neurospora crassa,
we started with the creation of a microarray prior to sequence assembly
and
annotation, however. Some 4,700 EST-clones were arrayed on glass slides
and
used to monitor nutrient-dependent functional phenomena in Neurospora
crassa. Upon availability of the sequence, also arrays made by in situ synthesis of
oligonucleotides were used in other analyses.
...
Aign & Hoheisel (2003) Fungal
Genet. Biol. 40, 225-233.
.
|

| Vingron & Hoheisel |
|
(1999) |
|
J. Mol. Med. 77, 3-7. |
|
| Beissbarth et al. |
|
(2000) |
|
Bioinformatics 16,
1014-1022. |
|
| Fellenberg et al. |
|
(2001) |
|
Proc. Natl. Acad. Sci.
USA 98, 10781-10786. |
|
| Hoheisel et al. |
|
(2001) |
|
Bioforum 12/01,
908-910. |
|
| Fellenberg et al. |
|
(2002) |
|
Bioinformatics 18,
423-433. |
|
| Brazma et al. |
|
(2002) |
|
Adv. Biochem.
Biotechnol. 77, 113-139. |
|
| Fellenberg et al. |
|
(2003) |
|
Perspectives in Gene
Expression, Eaton Publishing,
Westborough, 307-343. |
|
| Busold et al. |
|
(2005) |
|
Bioinformatics
21, 2424-2429. |
|
| Fellenberg et al. |
|
(2006) |
|
BMC Genomics 7, 319.
|
|
| Moghaddas Gholami & Fellenberg |
|
(2010) |
|
Bioinformatics 26, 1082-1090. |
|
-----------------------------------------------------------------
other publications
and patents
|
|
|
..
FINISHED
PROJECT:
Multi-Conditional
Hybridisation Intensity Processing System (M-CHiPS)
Initial
data analysis software
tools and an appropriately structured database were developed by Kurt
Fellenberg in close
collaboration with Martin
Vingron and went live in 1999. From this
work resulted the M-CHiPS data warehouse
and analysis software package, which
was designed and implemented by Kurt
Fellenberg. Apart from our own projects, the package is being
used
by various external partners and other groups elsewhere.
..
Eventually,
the data warehouse held
results of more than 13,000
experiments.
..
The Multi-Conditional
Hybridisation Intensity Processing System (M-CHiPS)
is a data warehouse, which provides a structure suitable for
statistical analysis of a microarray database's entire content,
including components such as the experimental and clinical annotations,
for example. The storage concept is flexible and accounts for future
developments. For each organism, there is a specific database. Although
these databases may contain different ontologies of experimental and
other annotations, they share the same structure and therefore can be
accessed by the very same statistical algorithms. An
ontology-independent structure enables ontology-updates during normal
database operation, avoiding structure-alterations.
..
For
overall data analyses as well as the identification of associations
between transcriptional variations and annotated factors, including
clinical information, GO-terms, mapping data and such alike,
correspondence
analysis is used extensively. It is an explorative computational method
for the study of associations between variables and proved its
usefulness for identifying factors, which are associated to certain
phenotypes, for example. Much like principle component analysis, it
displays a low-dimensional projection of the overall data matrix. One
major advantage of the process is its ability to present different
parameters of a multi-dimensional data matrix (e.g., genes and
experimental conditions) in a single plot. Localisation of genes and
individual
experiments
is an indicator for an association between them. Moreover, additional
information, such as GO-term or clinical
annotations, can be displayed
also, permitting an immediate identification of regulated functional
groups or pathways). In addition,
algorithms have been established to identify from the annotated data
the factors, which are likely to be causative for the establishment of
certain sub-groups (clusters) of factors (e.g., genes or patient
groups) by statistical data evaluation.
|
|