Functional
Genome Analysis (B070)
Deutsches
Krebsforschungszentrum,
Im Neuenheimer Feld 580
D-69120
Heidelberg,
Germany. |
 |
..
.
..
Epigenetics
..
..
..
..
..
|
DNA-methylation studies
Changes
in genomic DNA methylation patterns are early and consistent
features of tumourigenesis. Aberrant DNA
methylation profiles can thus be used as a valuable markers for
clinical
tumour
characterisation. Technically, we
initially applied
microarray
technology toward a genome-wide and high-resolution
analysis of DNA
methylation
patterns. Meanwhile, next-generation
sequencing
and other techniques are used.
...
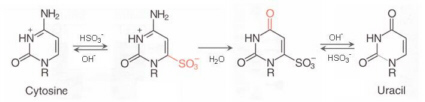
The
detection of
methylation
variations is performed by using bisulfite
treatment to
uncover
the methylation status. Sodium bisulfite induces methylation-dependent
single-nucleotide polymorphisms by converting unmethylated cytosine to
uracil
and,
upon PCR amplification, to thymine (see figure below).
5-Methylcytosine is not affected by
sodium
bisulfite treatment and thus amplified as cytosine. The
conversion can be identified by any means of sequence analysis.
The data are evaluated in
combination with available
clinical data
and information from other analyses, such as transcript analyses.
This allows
insights into the role of DNA methylation during tumourigenesis. We study in some detail
the functional consequences of variations
of particularly promoter methylation, and have uncovered
relevant functional mechanisms in several cancer
entities.
|
|
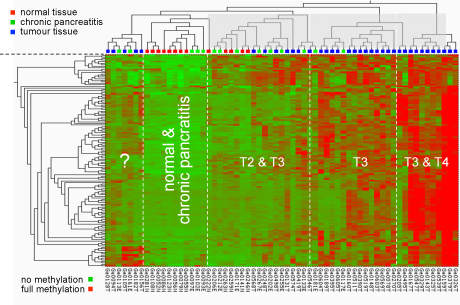
Early analysis results:
Methylation pattern at CpG dimers in promoters of cancer-relevant
genes. The type of pancreas tissue
analysed is
shown at the top; the degree of methylation is indicated by a
colour-code ranging from green (no methylation) to red
(hypermethylation).
|
|
|
|
|
|
|
|
|
Bermejo et al. (2019) Epigenomics 11, 81. |
 |
|
|
|
|
|
|
|
Wu et
al.
(2021) Cancers 13, 4569. |
|
|
Bure et al. (2018)
Genes Chrom. Cancer
57, 584. |
|
|
Bubnov et al. (2012) Exp.
Oncol. 34, 370. |
 |
|
|
|
|
Manoocherhri et al. (2021) Int. J. Cancer 52, 1025-1035. |
|
|
Meder et al. (2017) Circulation 136, 1528. |
|
|
Botla et
al. (2012) Breast Canc. Res.
Treat. 135, 705. |
|
|
|
|
|
Manoocherhri et al. (2021) Clin. Epigenetics 13, 207. |
|
|
Botla et al. (2016) Cancer Res. 76, 4149. |
|
|
Moskalev et al. (2012) BMC Cancer 12, 213. |
|
|
|
|
|
Kapsner et al. (2021) Int. J. Cancer 149, 1150-1165. |
|
|
Moskalev et al. (2015) Oncotarget 6, 4418. |
|
|
Moskalev et al. (2012) Genes Chrom. Cancer 51, 105. |
|
|
|
|
|
Manoocherhri et al. (2020) Sci. Rep. 10, 11762. |
|
|
Jandaghi et al. (2015)
Cell Cycle 14, 689. |
|
|
Moskalev et al. (2011) Nucleic Acids Res. 39, e77. |
|
|
|
|
|
Manoocherhri et al. (2020) Mol. Oncol. 14, 1252-1267. |
|
|
Haller et al. (2014) Int. J. Cancer 136, 1013. |
|
|
de Souza Rocha Simonini et
al. (2010) Cancer
Res. 70, 9175. |
|
|
|
|
|
Visvanathan et al. (2019) Genes 10, 141. |
|
|
Dutruel et al. (2014) Oncogene 33, 3401. |
|
|
Böttcher et
al. (2010) PloS
ONE 5, e11002. |
|
|
|
|
|
Amini et al. (2019) J. Cell. Physiol. 234, 15320-15329. |
|
|
Haas et
al. (2013) EMBO
Mol. Med. 5, 413. |
|
|
Riazalhosseini &
Hoheisel (2008) Genome Biol.
9, 405. |
|
|
|
|
|
|
|
..
Epigenetic
signature that differentiates pancreatic cancer from chronic
pancreatitis
.
Diagnostic differentiation between PDAC and
chronic pancreatitis (CP) is challenging and currently only 65% accurate in clinical practice. Misdiagnoses
in both directions have severe consequences for the patients. We set
out to
define molecular markers for a clear distinction of PDAC and CP. We established a random-forest based
machine-learning approach to identify markers for patient
classification
comparing the performance of data from genome-wide DNA methylation, mRNA and miRNA transcriptional analyses as well as combinations
thereof. The approach succeeded in defining accurate
markers, while low-dimensional embedding and cluster
analysis failed to do so. Variations in DNA methylation yielded the best diagnostic accuracy, dwarfing the
importance of
transcript levels. Identified changes were validated with data taken
from
public repositories and confirmed in an independent sample set. A signature made of few DNA methylation sites
achieved a validated diagnostic accuracy of 100%, which even included
some
degree of redundancy for diagnostic robustness.
The success of the
machine-learning process to identify a highly effective marker
signature
documents the power of this approach. The substantially improved
diagnostic
accuracy in patients with suspected pancreatic cancer could have
tremendous
consequences for patient management.
|
..
..
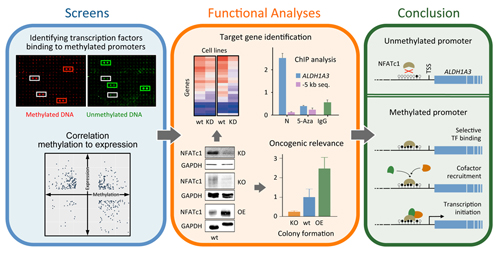
Promoter methylation promotes the
binding of transcrption factor NFATc1, triggering oncogenic gene
activation in pancreatic cancer
Studies
have indicated that some genes involved in carcinogenesis are highly
methylated
in their promoter regions but nevertheless strongly transcribed. It has
been
proposed that transcription factors could bind specifically to
methylated
promoters and trigger transcription. We looked at this rather
comprehensively
for pancreatic ductal adenocarcinoma (PDAC) and studied some cases in
more
detail. Some 2% of regulated genes in PDAC exhibited higher
transcription
coupled to promoter hypermethylation in comparison to healthy tissue.
Screening
661 transcription factors, several were found to bind specifically to
methylated promoters, in particular molecules of the NFAT family. One
of them -
NFATc1 - was substantially more expressed in
PDAC than control tissue and exhibited a strong oncogenic role.
Functional
studies combined with computational analyses allowed determining
affected
genes.
A prominent one was gene ALDH1A3, which accelerates PDAC
metastasis and correlates with
a bad prognosis. Further studies confirmed the direct
up-regulation of ALDH1A3
transcription by NFATc1 promoter binding in a methylation-dependent
process,
providing insights into the oncogenic role of transcription activation
in PDAC
that is promoted by DNA methylation.
Wu et
al.
(2021) Cancers 13, 4569.
|
|
|
.
|
..
|
|
|
SST hypermethylation acts as a
pan-cancer marker
Aberrant DNA methylation is often involved in carcinogenesis. A
genome-wide methylation study was performed on DNA from pancreatic
ductal
adenocarcinoma (PDAC) and endocrine pancreas tumours. Validation of DNA
methylation patterns and concomitant alterations in expression of gene
candidates was performed on patient samples and pancreatic cancer cell
lines.
Furthermore, validation was done on independent data from The Cancer
Genome
Atlas (TCGA) and Gene Expression Omnibus (GEO). Finally, droplet
digital PCR was
employed to detect DNA methylation marks in cell-free (cf) DNA isolated
from
plasma samples of PDAC patients and cancer-free blood donors.
.
Hypermethylation of the SST
gene (encoding somatostatin) and concomitant down-regulation of its
expression
was discovered in PDAC and endocrine tumour tissues while not being
present in
chronic pancreatitis (inflamed) tissues and normal pancreas. Fittingly,
treatment with a somatostatin agonist (Octreotide) reduced cell
proliferation
and migration of pancreatic cancer cells. Diagnostic performance of SST methylation in a receiver operating
characteristic (ROC) curve analysis was 100% and 89% for tissue and
plasma
samples, respectively.
.
A large body of TCGA and GEO data confirmed SST
hypermethylation and down-regulation
in PDAC and showed a similar effect in a broad spectrum of other tumour
entities. SST promoter methylation
represents a sensitive and promising molecular, pan-cancer biomarker
detectable
in tumour tissue and liquid biopsy samples..
Manoocherhri et
al. (2020) Mol.
Oncol. 14, 1252-1267.
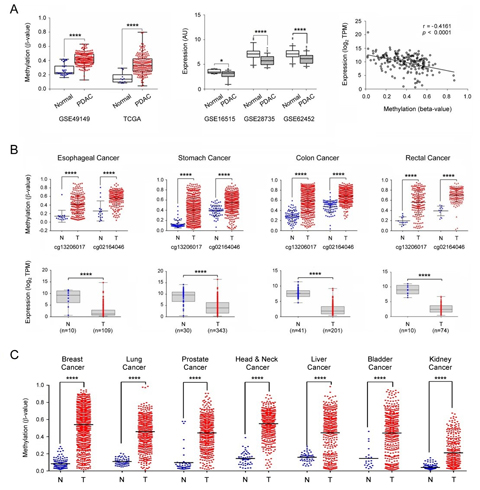
Figure legend: External validation of SST expression and
methylation. (A) The SST gene
methylation was looked at in two independent datasets about pancreatic
cancer
obtained from GEO and TCGA (left panel). The central panel shows the
down-regulation of SST gene expression
in PDAC in three independent datasets from GEO. In the right panel, the
Pearson
correlation of DNA methylation and gene expression levels of the SST gene are shown (data from
TCGA-PAAD). (B) Investigation of SST
gene methylation and expression
in esophageal, stomach, colon, and rectal adenocarcinomas. The β values
of two
probes that were common to the 450k and 27k methylation arrays were
applied for
comparison. (C) SST gene
methylation results are shown
as in panel B but for breast, lung, prostate, head & neck, liver,
bladder
and kidney cancer. TPM: transcripts per million; *: p ≤ 0.05; ****: p ≤
0.0001.
|
|
.
|
..
|
M6-Methyladenoadenosine landscape of glioma stem-like cells: METTL3 is
essential for the expression of actively transcribed genes and
sustenance of the oncogenic signalling
Despite
recent advances in m6A biology,
the regulation of crucial RNA processing steps by the RNA methylase
METTL3 in
glioma stem-like cells (GSCs) remains obscure. An integrated analysis
of m6A-RNA-immunoprecipitation
and total RNA-sequencing of METTL3-silenced GSCs identified that m6A
modification in GSCs is principally carried out by METTL3. The m6A-modified
transcripts showed higher abundance compared to non-modified
transcripts.
Further, METTL3 is essential for the expression of
GSC-specific actively transcribed genes. Silencing METTL3 resulted in
an
elevation of several aberrant, alternative splicing events. Putative m6A
reader proteins play a key role in the RNA
stabilization function of METTL3. METTL3 altered A-to-I and C-to-U RNA
editing
events by differentially regulating RNA editing enzymes ADAR and
APOBEC3A.
Similar to protein-encoding genes, lincRNAs with m6A-marks
showed
METTL3-dependent high expression. m6A modification of
3’-UTRs
appears to result in a conformation-dependent hindrance of miRNA
binding to
their targets. The integrated analysis of the m6A regulome
in
METTL3-silenced GSCs showed global disruption in tumorigenic pathways
that are
indispensable for GSC maintenance and glioma progression. We conclude
that
METTL3 plays a vital role in many steps of RNA processing and
orchestrates
successful execution of oncogenic pathways in GSCs.
Visvanathan et al. (2019) Genes 10,
141.
|
.
..
..
..
|
Methylation in the gene encoding the
long intergenic non-coding RNA 299 serves as biomarker in peripheral blood for triple-negative breast cancer
Worldwide,
breast cancer was the most frequently diagnosed cancer and cause of
cancer
death among women in 2012.
Triple-negative breast cancer (TNBC), defined by lack of (i) tumour
expression
of estrogen receptor (ER), (ii) progesterone receptor (PR), and (iii)
human epidermal growth factor receptor 2
(HER2), is the most aggressive subtype of breast
cancer and accounts for 10% to 20% of all diagnoses. We screened
the epigenome-wide
methylation profiles of 233 TNBC patients and 233 age-matched controls
(discovery
set) and validated the most promising methylation probes in 57 TNBC
patients
and 124 controls (validation set) with the aim of identifying
epigenetic biomarkers based on DNA
from peripheral blood leukocytes.
..
..
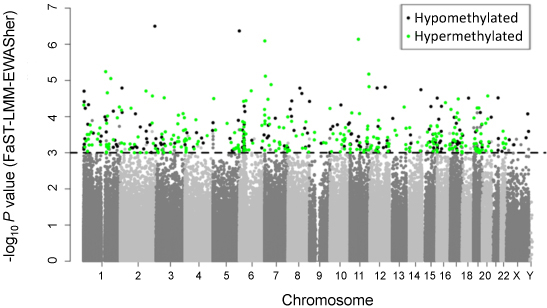
Figure legend: Manhattan
plot showing the chromosomal distribution of the p-values of 370,706
methylation variations in TNBC. The dashed line indicates the 0.001
significance threshold.
..
..
Bermejo et al. (2019) Epigenomics 11, 81.
|
|
|
|
..
..
|