Functional
Genome Analysis (B070)
Deutsches
Krebsforschungszentrum,
Im Neuenheimer Feld 580
D-69120
Heidelberg,
Germany. |
 |
..
.
..
Functional
Tumour Analyses / Pancreatic Cancer..
..
..
Pancreatic cancer
Pancreatic
ductal
adenocarcinoma (PDAC) is the currently most deadly cancer. Mortality is
close to incidence. Only about 5% of all patients survive longer than
five years. Untreated, the average survival period
after diagnosis is about five months. Surgical
resection represents still the best curative treatment approach for
pancreatic cancer. However, it
can
only be applied to 10% to 20% of patients. PDAC is
currently the fourth and seventh most common cause of cancer-related
death in the Western world (Europe and the USA) and China,
respectively, although ranked only tenth in incidence. Numbers are
continuously rising, however, while declining for many other
tumour entities.
In 2030, PDAC is expected to be the second most cause of cancer-related
death in the Western world, surpassing colorectal and breast cancer.
...
Joining
forces with leading groups in basic and clinical pancreatic cancer
research, we study
various molecular aspects in order to
improve the basic understanding of PDAC tumour biology and pathology. To
this end, we look in much detail at the communication between
different cell types within the tumour microenvironment, for example,
with a special
focus on proteins. All this is
complemented by studies on the functional
consequences of the observed molecular
changes.
...
Taking advantage of the gained knowledge, we are aiming
at establishing novel approaches for
early diagnosis, disease prognosis and risk stratification of
pancreatic
cancer. To this end, we pursue particularly non-invasive liquid
biopsy processes based on blood samples. Applying and combining
different analysis forms, molecular signatures at various
levels are defined that could enable accurate diagnosis.
...
Also, we are
active in identifying new therapeutic routes of high potential so as to
improve on the current prognosis by means of innovative
therapeutic strategies. One
possible therapeutic option is at a stage that requires a clinical
trial as the next step in the evaluation of its clinical utility.
...
All this is done in several collaborations
with partners worldwide. We have
participated repeatedly in
international and national consortia that covered various
aspects
of pancreatic cancer research. Locally, we
have a strong
and continuous cooperation with the European
Pancreas Center (EPZ) at the Surgery
Department of the Heidelberg University Hospital.
|
|
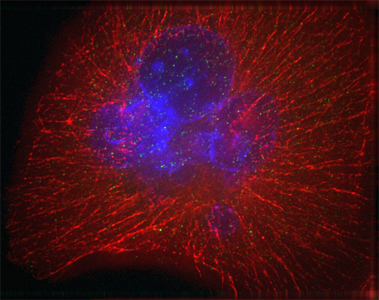
...
Image of a pancreatic cancer cell.
DNA in the nucleus is labelled blue; red signals show the cytoskeleton.
|
|
|
|
|
|
|
Njouendou et al.
(2023) Mol. Cancer 22, 52. |
 |
|
Stoy et
al. (2015) EMBO Mol. Med. 7,
1048-1062. |
 |
|
Youns et
al. (2009) Biochem.
Pharmacol. 78, 273-283. |
|
|
|
|
|
Zhang et al.
(2023) Clin. Cancer
Res. 29, 1535-1545. |
|
|
Jandaghi et al. (2015) Cell Cycle 14,
689-690. |
|
|
Ketterer et
al. (2009) Cancer Lett.
277,
72-81. |
|
|
|
|
|
Heid et
al.
(2022) Cancer Metab. 10, 24. |
|
|
Moskalev et al.
(2015) Oncotarget 6,
4418-4427. |
|
|
Börner et
al. (2009) Biotechniques 46,
297-304. |
|
|
|
|
|
Brindl et al.
(2022) Cancers 14, 3562. |
|
|
Haller et al. (2015) Int. J.
Cancer 136, 1013-1023. |
|
|
Bauer et
al. (2009) Pancreatology 9,
34-44. |
|
|
|
|
|
Wu et
al.
(2021) Cancers 13, 4569. |
|
|
Keller et al. (2014) BMC Med. 12,
224. |
|
|
Loos et al. (2007) Virchows Arch. 450,
719-726. |
|
|
|
|
|
Al-Shaheri et
al. (2021) Cancer Treat. Rev. 96,
102193. |
|
|
Keleg et al. (2014) PLoS ONE
9,
e100178. |
|
|
Bauer et
al.
(2007) Grundl.
Mol. Med., Springer, 346-362. |
|
|
|
|
|
Roth et
al. (2021) Annals Surgery 273,
e273-e275. |
|
|
Dutruel et al. (2014) Oncogene
33,
3401-3410.7. |
|
|
Kusumawidjaja
et al. (2007) Canc. Biol.
Ther. 6, 367-376. |
|
|
|
|
|
Ghassem-Zadeh et
al. (2020) Int.
J. Mol. Sci. 21, 2403. |
|
|
Wolf et al. (2013) PLoS ONE
8,
e74555. |
|
|
Jesnowski et
al.
(2006) Neoplesia 9,
136-146. |
|
|
|
|
|
Manoochehri et
al. (2020) Mol. Oncol. 14,
1252-1267. |
|
|
Youns &
Fathy (2013) J. Cell.
Biochem. 114, 2654-2665. |
|
|
Fellenberg et
al. (2006) BMC Genomics 7,
319. |
|
|
|
|
|
Al Alawi et al. (2020) Biomed.
Pharmacother. 121, 109522. |
|
|
Fredebohm et al. (2013) J. Cell Sci. 126,
3380-3389. |
|
|
Buchholz et
al.
(2005) Clin. Cancer
Res. 11, 8048-8054. |
|
|
|
|
|
Miao et
al. (2020) Int. J. Cancer 147,
189-201. |
|
|
Campa et al. (2013) Can.
Epid. Biom. Prev. 22, 320-322. |
|
|
Buchholz et
al. (2005) Exocrine
Panc. Cancer,
Solvay,
396-403. |
|
|
|
|
|
Ali et
al.
(2019) Cell Death Disc. 5,
128. |
|
|
Rachakonda et al. (2013) PLoS ONE
8,
e60870. |
|
|
Beier et
al. (2005) Exocrine
Pancreas Cancer, Solvay,
254-260. |
|
|
|
|
|
Bakadlag et al. (2019) Expert Opin. Ther. Tar. 23, 365-367. |
|
|
Marzoq et
al. (2013) J. Biol.
Chem. 288,
32517-32527. |
|
|
Busold et
al. (2005) Bioinformatics 21,
2424-2429. |
|
|
|
|
|
Marzoq
et
al.
(2019) Sci. Rep. 9, 5303. |
|
|
Rizzato et al.
(2013) Oncol. Report 29, 1637-1644. |
|
|
Brandt et
al.
(2004) Pancreatology 4,
587-597. |
|
|
|
|
|
Pausch
et
al.
(2018) Pancreas 47,
561-567. |
|
|
Hoheisel et
al. (2013) Proteomics
Clin. Appl. 7, 8-16. |
|
|
Esposito et
al. (2004) Virchows
Arch. 444,
447-453. |
|
|
|
|
|
Youns et
al.
(2018)
Naunyn-Schmiedebergs 391, 551-560. |
|
|
Fredebohm et al. (2012) PLoS ONE 7,
e48503. |
|
|
Hoheisel, J.D. (2003) Forschung
&
Diagnostik 1, 34-35. |
|
|
|
|
|
Throm
et
al.
(2018) Oncotarget 9, 11734-11751. |
|
|
Alhamdani et
al. (2012) J. Proteomics 75, 3747-3759. |
|
|
Bauer et
al. (2003) Comp. Funct.
Genom. 4,
520-524. |
|
|
|
|
|
Bauer
et
al.
(2018) Int. J.
Cancer 142,
1010-1021. |
|
|
Bauer et al. (2012) PLoS ONE 7, e34151. |
|
|
Löhr
& Hoheisel (2003) Z.
Gastroenterol. 41,
623-624. |
|
|
|
|
|
Kaistha et
al.
(2017) Oncotarget
8,
66215-66225. |
|
|
Rizzato et al. (2011) PLoS ONE
6,
e27921. |
|
|
Frohme et
al. (2000) Mol. Pathog.
Pancreatic
Cancer, IOS Press, 88-94.
|
|
|
|
|
Mustafa et
al.
(2017) Oncotarget
8,
11963-11976. |
|
|
Keller et al. (2011) Nature
Meth. 8,
841-843. |
|
|
Geng et al.
(1999) Expression
Genetics, Biotechniques Books, 145-153.
|
|
|
|
|
Jandaghi et
al.
(2016) Gastroenterology 151,
1218-1231. |
|
|
Holtrup et al. (2011) Brit. J.
Pharmacol. 162, 1045-1059. |
|
|
Wallrapp et
al.
(1999) Ann. Oncology 10,
suppl. 4, 64-68. |
|
|
|
|
|
Botla et al.
(2016) Cancer Res. 76,
4149-4159. |
|
|
Youns et al. (2011) Eur. J.
Pharmacol. 650, 170-177. |
|
|
Wallrapp et
al.
(1999) Ann. N. Y.
Sci. 880, 122-146. |
|
|
|
|
|
Cheng et al.
(2016) Mol. Oncol. 10, 806-82. |
|
|
Schröder et
al. (2010) Mol. Cell.
Prot. 9,
1271-1270. |
|
|
Geng et al.
(1998) Biotechniques 25,
434-438. |
|
|
|
|
|
Heller et al. (2015) Pancreatology 15,
423-431. |
|
|
Alhamdani et
al. (2010) J. Prot. Res. 9,
963-971. |
|
|
Gress et
al.
(1997) Genes Chrom.
Cancer 19, 97-103. |
|
|
|
|
|
|
|
.
Blood-based
diagnosis and risk stratification of patients with intraductal
papillary mucinous neoplasm (IPMN) to decide on surgical intervention
Intraductal
papillary mucinous neoplasm (IPMN) is a
precursor of PDAC. Patients with low-grade dysplasia have a relatively
good
prognosis and are kept under surveillance to monitor disease
development,
whereas high-grade dysplasia and IPMN invasive carcinoma require tumour
resection. Diagnostic distinction of the two groups is difficult,
however. We
aimed to identify variations in protein concentration in peripheral
blood for
accurate discrimination. Sera from IPMN patients and healthy donors were
analysed on microarrays made of 2,977
antibodies. For microRNA biomarkers, a PCR-based screen was performed
and
biomarker candidates confirmed by quantitative PCR.
A support vector
machine
(SVM) algorithm defined classifiers, which were validated on a separate
sample
set. A panel of five proteins and three miRNAs could distinguish high-
and
low-risk IPMN with an accuracy of 97%. This is substantially better
than the
accuracy obtained in the same patient cohort by using the guideline
criteria
for decision-making on performing surgery or not. The precise
blood-based
diagnosis and risk stratification will improve patient management and
thus the
prognosis of IPMN patients. In addition to the main finding, highly
accurate
discrimination was also achieved between other patient subgroups.
Zhang
et
al.
(2023) Clin. Cancer
Res. 29, 1535-1545.
|
|
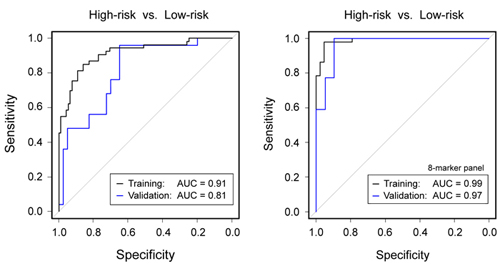
Figure legend: (Left) Diagnostic
performance of
clinical parameters to discriminate high-risk from low-risk IPMN
according to
current guidelines. (Right) Much
better results were obtained by a combined panel of 8 protein and miRNA
biomarkers. The results are presented as ROC curves and corresponding
AUC
values as determined in the training and validation cohorts,
respectively. |
.
|
.
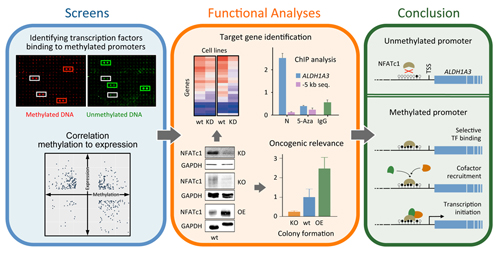
Promoter methylation promotes the
binding of transcrption factor NFATc1, triggering oncogenic gene
activation in pancreatic cancer
Studies
have indicated that some genes involved in carcinogenesis are highly
methylated
in their promoter regions but nevertheless strongly transcribed. It has
been
proposed that transcription factors could bind specifically to
methylated
promoters and trigger transcription. We looked at this rather
comprehensively
for pancreatic ductal adenocarcinoma (PDAC) and studied some cases in
more
detail. Some 2% of regulated genes in PDAC exhibited higher
transcription
coupled to promoter hypermethylation in comparison to healthy tissue.
Screening
661 transcription factors, several were found to bind specifically to
methylated promoters, in particular molecules of the NFAT family. One
of them -
NFATc1 - was substantially more expressed in
PDAC than control tissue and exhibited a strong oncogenic role.
Functional
studies combined with computational analyses allowed determining
affected
genes.
A prominent one was gene ALDH1A3, which accelerates PDAC
metastasis and correlates with
a bad prognosis. Further studies confirmed the direct
up-regulation of ALDH1A3
transcription by NFATc1 promoter binding in a methylation-dependent
process,
providing insights into the oncogenic role of transcription activation
in PDAC
that is promoted by DNA methylation.
Wu et
al.
(2021) Cancers 13, 4569.
|
|
|
.
|
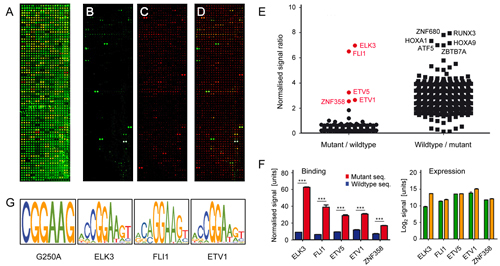
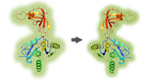
Figure
legend: Identification
of transcription factors that bind preferentially to
the mutant hTERT promoter. (A) A
protein microarray is shown that presents
667
transcription factor DNA binding domains. Proteins were immunostained
with
fluorescently labelled antibodies that target terminal tags. Incubation
of a 35
bp fragment resembling (B) the
mutated or (C) the wildtype version
of the promoter sequence identified specific binding; in (D)
a merger of the images is shown. (E) The ratio,
resulting from four independent experiments, is
presented of the normalized binding signals. Transcription factors are
highlighted in red, which exhibited stronger binding to the mutated
sequence. (F) The results of few transcription
factors are presented in more detail (left panel). In addition, the
relative
protein level was deduced from the antibody labelling of their terminal
tags
(right panel). Green and orange bars indicate the signal intensities
obtained
at the N- and C-termini, respectively (G)
Sequence recognition motifs of the three best binding transcription
factors are
depicted in comparison to the binding motif generated by the mutations
(leftmost panel).
|
|
Transcription
factor FLI1 promotes cancer progression by affecting cell cycle
regulation
Binding of
transcription factors to mutated DNA
sequences is a likely regulator of cancer progression. Noncoding
regulatory
mutations such as those on the core promoter of the gene encoding human
telomerase reverse transcriptase have been shown to affect gene
expression in
cancer. Using a protein microarray
of 667 transcription factor DNA-binding domains and subsequent
functional
assays, we looked for transcription factors that preferentially bind
the mutant
hTERT promoter and characterized
their downstream effects.
One of them, friend leukemia integration 1 (FLI1),
exhibited particularly strong effects with respect to regulating hTERT expression, while the even better
binding ELK3 did not. Depletion of FLI1 decreased expression of the
genes for
cyclin D1 (CCND1) and E2F
transcription factor 2 (E2F2)
resulting in a G1/S cell cycle arrest and in consequence a reduction of
cell
proliferation. FLI1 also affected CMTM7,
another gene involved in G1/S transition,
although by another process that suggests a balanced regulation of the
tumour
suppressor gene’s activity via opposing regulation processes.
FLI1 expression was found upregulated and
correlated with an increase in CCND1
expression in pancreatic cancer and brain tumours. In non-neoplastic
lung
cells, however, FLI1 depletion led to rapid progression through the
cell cycle.
This coincides with the fact that FLI1 is down-regulated in lung
tumours. Taken
together, our data indicate a cell cycle regulatory hub involving FLI1,
hTERT, CCND1 and E2F2 in
a
tissue- and context-dependent manner.
Miao
et
al.
(2020) Int. J.
Cancer 147, 189-201. |
..
..
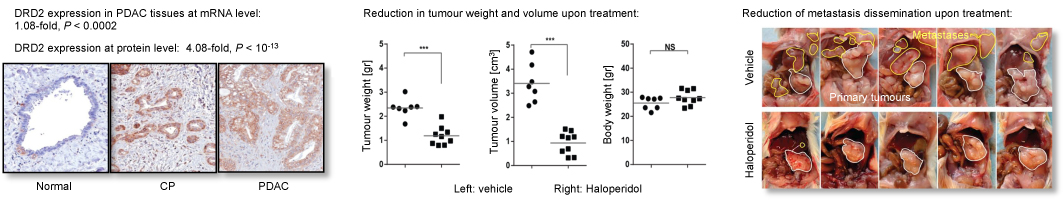
Novel
Therapy Option:
DRD2 is
critical for pancreatic cancer and promises pharmacological therapy by
already established antagonistsr
Incidence and
mortality of pancreatic ductal adenocarcinoma (PDAC) are almost
equivalent,
urging for the development of better therapeutic strategies. We
investigated novel
potential therapeutic targets for PDAC
by performing global gene
expression profiling in 195 PDAC and 41 normal pancreatic tissue
samples. Superimposing
the pathway context and interaction networks of aberrantly expressed
genes, we
identified factors with central roles
in PDAC pathways. Next, tissue microarray analysis was used to
verify
the expression of the candidate targets in an independent set of 152
samples
comprising 40 normal pancreatic tissues, 63 PDAC sections and 49
samples of chronic
pancreatitis.
We identified dopamine receptor D2 (DRD2) as a key modulator of
cancer pathways in PDAC. DRD2 up-regulation at the protein level was
validated
in a large independent sample cohort. Most importantly, we
found that
blockade of DRD2, through RNAi or pharmacological inhibition using
FDA-approved
antagonists, such as Haloperidol, hampers the proliferative and invasive
capacities of pancreatic cancer cells in
vitro and in vivo while
modulating cAMP and endoplasmic reticulum stress pathways. Our findings
demonstrate that inhibiting DRD2 represents a novel therapeutic
approach for
PDAC. Given that DRD2 antagonists are currently routinely used for the
management of schizophrenia or other mental diseases, a
drug-repositioning strategy could
facilitate the
clinical use of these agents for treating pancreatic cancer.
.
Bakadlag et al. (2019) Expert Opin. Ther. Tar. 23, 365-367.
Jandaghi et al. (2016) Gastroenterology 151,
1218-1231.
|
..
|