|
|
|
|
|
|
|
|
|
|
|
|
|
|
Functional
Genome Analysis (B070)
Deutsches
Krebsforschungszentrum,
Im Neuenheimer Feld 580
D-69120
Heidelberg,
Germany. |

|
|
|
|
|
|
|
|
|
|
|
|
..
..
For de novo
large-scale sequencing projects, the availability of physical maps is
desirable. The existence of a reliable clone map is extremely helpful
even in
shotgun sequencing projects. All of them have taken into account some
sort of
mapping information for contig alignment and control of colinearity.
Based on
earlier results on Drosophila melanogaster, Schizosaccharomyces
pombe,
Saccharomyces cerevisiae, Arabidopsis thaliana, Trypanosoma cruzi
and
human, mapping projects were pursued to provide a scaffold for
subsequent or
parallel sequence analysis or directly for the preparation of probe
molecules
for functional studies.
In the
projects on Pseudomonas
putida, for example, the shotgun clones were immediately used for
transcriptional profiling analysis. To this end, a minimal tiling path
was
identified and placed on microarrays in form of PCR-products. Since
this
microarray does exhibit all coding regions of the genome, transcript
analyses
are performed by definition on a complete gene representation of the
organism,
irrespective of the status of the sequence annotation. Similar work was
done in
other projects, such as studies on Trypanosoma brucei and Drosophila
melanogaster.
|
 |
|
|
|
FINISHED
PROJECT:
Directed
gap closure in large-scale sequencing projects
A
problem in many
sequencing projects is the final closure of gaps left in the clone
libraries,
which serve as templates for sequencing, because of uncloned or
unclonable
genomic areas. Using the Xylella fastidiosa genome as a test
system, a
technique was established to generate in a directed manner sequence
information
from those gaps. We used the complete clone library as a competitor
against the
genomic DNA of interest in a subtractive hybridisation procedure
similar to
representational difference analysis (RDA). The resulting sequence
information
serves directly for gap closure or can be used to screen selectively
other
clone resources.
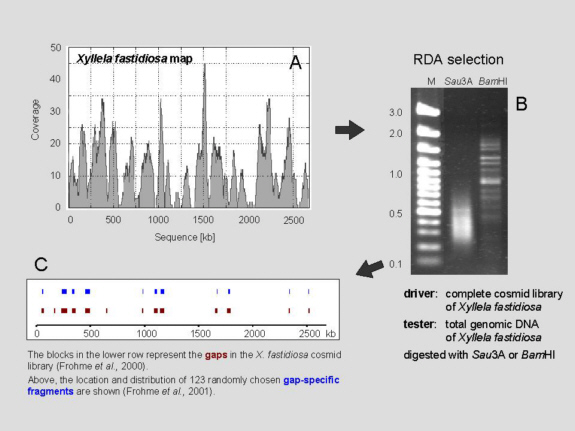
Figure.
(A)
Variance in genome coverage of clone library.
(B)
Gel separation of the difference products from a comparison of genomic
DNA versus library DNA. The enzymes Sau3AI and BamHI
had been
used for the initial restriction digests; M: 100 bp marker ladder.
(C)
Individual fragments were picked at random and end-sequenced. 74% of
the sequences were not contained in the clone library but in the final X.
fastidiosa genome sequence. The relatively high portion of false
positives
is probably the result of the single step of subtraction enrichment,
done in an
attempt to reach a compromise between specificity and representation.
Frohme et al.
(2001) Genome Res. 11, 901-903.
.
|
 |
|
FINISHED
PROJECT:
Physical
mapping and transcriptional profiling analyses of the 6.1 Mb genome of Pseudomonas
putida.
In a network
made up by the
Medizinische Hochschule Hannover, the GBF in Braunschweig, QIAGEN in Hilden and us -
and in
collaboration with TIGR (U.S.A.) - the
entire genome of P. putida was sequenced. Within this network,
we
provided a physical clone map and analysed transcriptional
changes and genomic
differences between strains using microarrays produced from a minimal
tiling
path of shotgun sequencing clones.
Nelson et al. (2002) Environ.
Microbiol. 4, 799-808.
Stjepandic et al.
(2002) Environ. Microbiol. 4, 819-823.
Reva et al. (2006) J. Bacteriol. 188,
4079-4092.
|
FINISHED PROJECT:
Mapping
and sequencing of the Xyllela
fastidiosa genome.
. .
.
The
complete genome sequence
of X. fastidiosaclone 9a5c, which causes citrus variegated
chlorosis – a
serious disease of orange trees, was mapped and sequenced. Our
contribution was
the provision of a clone map used for both directed sequencing and as a
scaffold. The genome comprises a 52.7% GC-rich 2,679,305-base-pair (bp)
circular chromosome and two plasmids of 51,158 bp and 1,285 bp.
Putative
functions to 47% of the 2,904 predicted coding regions could be
assigned. The
mechanisms associated with pathogenicity and virulence involve toxins,
antibiotics and ion sequestration systems, as well as
bacterium–bacterium and
bacterium–host interactions mediated by a range of proteins.
Orthologues of
some of these proteins have only been identified in animal and human
pathogens;
their presence in X. fastidiosa indicates that the molecular
basis for
bacterial pathogenicity is both conserved and independent of host.
| Simpson et al. |
(2000) .
Nature 406, 151-157. |
|
| Frohme et al. |
(2000) .
Nucleic Acids Res. 28,
3100-3104. |
|
| Heber et al. |
(2000) .
Genomics 69, 235-241. |
|
| Heber et al. |
(2000) .J.
Comput. Biol. 7, 395-408. |
|
| Frohme et al. |
(2001) .Genome
Res. 11, 901-903. |
|
|
 |
|
|
 |
.
..
..
|

