Functional
Genome Analysis (B070)
Deutsches
Krebsforschungszentrum,
Im Neuenheimer Feld 580
D-69120
Heidelberg,
Germany. |
 |
..
.
..
Affinity
Proteomics
- Protein Microarrays
..
..
...
We
utilise protein
microarrays
containing (mostly full-length) molecules for the
investigation of protein-interactions.
Microarray production is
done by means of in situ
synthesis in a cell-free
transcription/translation process on the
microarrays, starting from
full-length cDNAs or PCR-products using gene-specific primers. Because of
two subsequent spotting events, no contamination is possible between
spots. Also, reagent consumption is kept to a minimum. Last, no
particular attachment process is required; expressed proteins stick to
their respective positions. Protein
interaction of all kinds as
well as the influence of co-factors, such as small molecules, on these events is
studied this
way. The complexity of the protein arrays produced so far ranges from a
few hundred molecules to some 14,000
individual proteins. The set-up
is used in various projects, frequently combining the obtained information
with other
data.
..
Personalised
proteomics:
In
a recently completed technical development, we added to the in situ protein production a
process that allows to present on the microarray the proteins in
exactly the conformation as they occur in tissues or other samples of
individual patients, reflecting all mutations or splice variants that
are specific for the particular sample. Thereby, the
effects of individual variations on protein interaction - with other
proteins, nucleic acids or small compounds - can be studied.
.
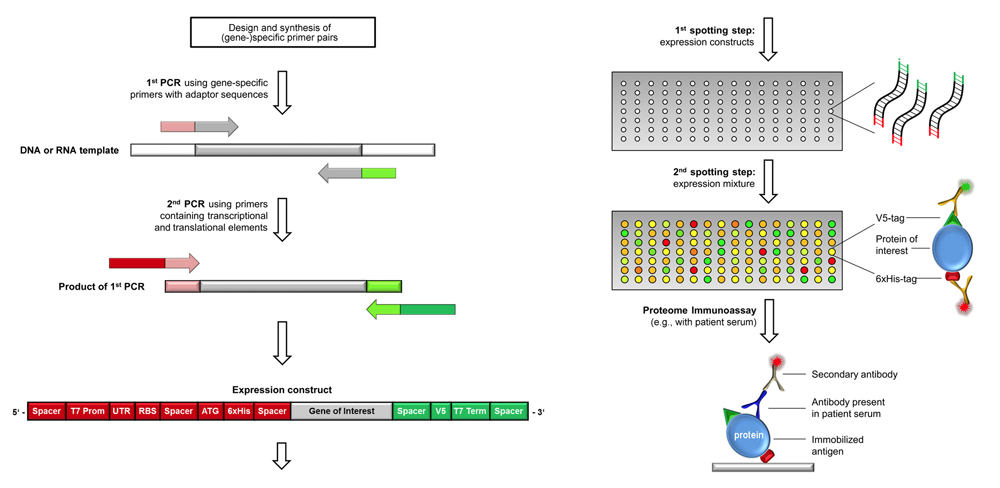
Scheme of producing complex proteome microarray by in situ
transcription & translation.
Testing for full-length products by using tag-specific antibodies
(shown on the right) is done for few, randomly selected arrays only.
|
|
|
|
|
Brindl et
al. (2022) Cancers 14, 3562. |
 |
|
Erben et
al. (2014) PLOS
Pathogens 10, e1004178. |
|
|
|
|
|
Ghassem-Zadeh et
al. (2020) Int. J. Mol.
Sci. 130, 2403. |
|
|
Syafrizayanti
et
al. (2014) Exp. Rev.
Prot. 11, 107. |
 |
|
|
|
|
Jeske et
al. (2020) Cancer
Epidem. Biomarkers Pref. 29,
2235-2242. |
|
|
Lueong et
al. (2013) J. Prot.
Bioinf. 07, 004. |
|
|
|
|
|
Hufnagel et
al. (2019) Bio-protocol 9, e3152. |
|
|
Hoheisel et
al. (2013) Proteomics
Clin. Appl. 7, 8. |
|
|
|
|
|
Hufnagel et
al. (2018) Sci. Rep. 8, 7503.
|
|
|
Schirwitz et
al. (2013) Adv. Materials 25, 1598-1602. |
|
|
|
|
|
Syafrizayanti et
al. (2017) Sci. Rep. 7,
39756. |
|
|
Schirwitz et
al. (2012)
Biointerphases 7, 47. |
|
|
|
|
|
Loeffler et
al. (2016) Nature Comm. 7,
11844. |
|
|
Friedrich et
al. (2011) Proteomics 11, 3757. |
|
|
|
|
|
Kibat et
al. (2016) New
Biotechnol. 33, 574-581.
|
|
|
Schmidt et
al. (2011) J. Prot. Res. 10, 1316. |
|
|
|
|
|
Lueong et
al. (2016) Mol. Microbiol. 100, 457-471. |
|
|
Gloriam et
al. (2010) Mol. Cell.
Prot. 9,
1. |
|
|
|
|
|
Betzen et
al. (2015) Proteomics Clin. Appl. 9, 342. |
|
|
Taussig et al. (2007) Nature
Meth. 4, 13. |
|
|
|
|
|
Mock et
al. (2015) Oncotarget 6, 13579-13590. |
|
|
Angenendt et al.
(2006) Mol.
Cell. Prot. 5,
1658. |
|
|
|
|
|
Hoheisel
(2014) labor&more 10/14, 10. |
|
|
Kersten et
al. (2005) Expert
Rev. Proteomics 2, 499. |
|
|
|
|
|
|
..
|
.
.
|
|
|
|
Identification of tumour-associated
autoantibodies and corresponding antigens in patient sera for
diagnostic purposes and the definition of novel therapy targets
Tumour-associated
antibodies are already present at early-stage cancer and circulate in
blood.
They could serve as a multiplier, yielding more circulating molecules
compared
with the corresponding antigens, even if the tumour is still small.
Based on
pilot studies with serum samples from patients with intraductal papillary mucinous neoplasm (IPMN; a
precursor of pancreatic
cancer), autoimmune
pancreatitis, chronic
pancreatitis (CP) and pancreatic
cancer (PDAC) (Ghassem
et al., 2020, Int. J. Mol. Sci.
21, 2403; Brindl et
al., 2022, Cancers 14, 3562), we studied the abundance of
autoantibodies on microarrays with up to 3060 human proteins (with an
emphasis
of cell surface proteins) in serum samples from healthy individuals and
patients with CP and all stages of IPMN and PDAC. Following an initial
screen,
1400 blood samples were studied in more detail. Data analysis has just
been
completed. Ninety-six biomarker candidates have also been checked for
their
capacity for early diagnosis by analysing by a Luminex immunoassay
thousands of
prospectively collected serum samples from patients, who in part
developed PDAC
at known time intervals after blood collection. The samples were made
available
by the US National Cancer Institute and studied in collaboration with
the DKFZ
division of Tim Waterboer.
Brindl et
al. (2022) Cancers 14, 3562.
Ghassem-Zadeh et
al. (2020) Int. J. Mol.
Sci. 130, 2403.
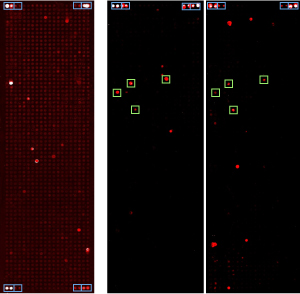
Figure legend: Protein microarrays are
shown that present some 1000 human proteins at full length. Incubation
was with antibodies isolated from patient sera. Red signals indicate
strong binding. Green frames highlight particular proteins that gave
rise to signals in several patients.
|
.
|
.
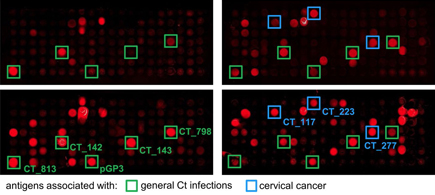
Immunoprofiling patients with
Chlamydia infection and cervical cancer
In a collaboration with the DKFZ group of Tim Waterboer, we describe a
method
for
identification of disease-related serum antibodies that are specific
for
infection and cancer, using Chlamydia
trachomatis (Ct) as a complex model organism. Bacterial
whole-proteome microarrays were
generated using
cell-free, on-chip protein expression. Expression constructs were
generated by
two successive PCR directly from bacterial genomic DNA. Bacterial
proteins
expressed on the microarray display antigenic epitopes, thereby
providing an
efficient method for immunoprofiling of patients. Antibodies from
patient serum was analysed as shown in the scheme above. Through
comparison of
antibody reactivity patterns, we identified antigens recognized by
known
Ct-seropositive samples, and antigens reacting only with samples from
cervical
cancer patients. Large-scale validation experiments using
high-throughput suspension bead array serology confirmed their
significance as
markers for either general Ct infection or cervical
cancer, providing
evidence for a role
of Ct infection in the development of cervical
cancer.
Hufnagel et
al. (2018) Sci. Rep.
8, 7503.
Hufnagel et
al. (2019) Bio-protocol 9, e3152.
|
|
|
.
Personalised
proteome analysis by means of
protein microarrays made from individual patient samples
|
DNA
sequencing has advanced to a state that permits
studying the genomes of individual patients as nearly a matter of
routine.
Towards analysing at a tissue’s protein content in a similar manner, we
established a method for the production of microarrays that represent
full-length proteins as they are encoded in individual specimens,
exhibiting
the particular variations, such as mutations or splice variations,
present in
these samples. From total RNA isolates, each transcript is copied to a
specific
location on the array by an on-chip polymerase elongation reaction,
followed by in situ cell-free transcription and
translation. These microarrays permit parallel analyses of variations
in
protein structure and interaction that are specific to particular
samples.
..
Apart from procedural information, details are provided on the overall
quality of protein microarrays (e.g., percentage of full-length
molecules and the structural integrity of proteins) and their
perfomance in various forms of analysis.
|
|
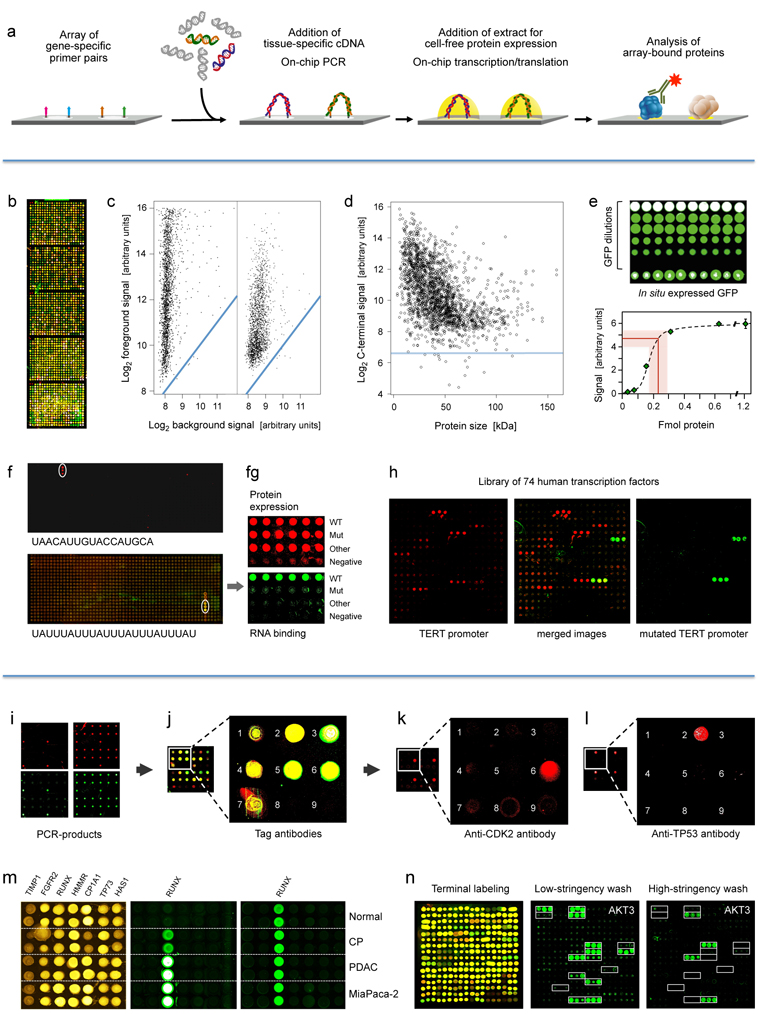 |
Figure.
..
First
panel: Principle and performance of the process of producing
personalised
protein microarrays. (a)
Schematic illustration of the overall process.
..
Second
panel: Protein microarray quality. (b)
Microarray with
2016 proteins that were expressed in situ;
visualisation was by incubation with red and green labelled antibodies
that
recognise common N- or C-terminal epitopes, respectively. (c)
The typical ratio is shown of foreground to background signals
for the N- (left) and C-terminus (right); blue lines indicate regions
of
identical intensity. (d) Detection
of the C-termini of the 2016 expressed proteins with Cy3-conjugated
anti-V5
antibody; the horizontal line represents a signal of three standard
deviations
above background. (e) Determination
of the amount of in situ synthesised
GFP by comparison to spotted material of known concentration; twenty
measurements each were done; the red line represents the average amount
of
synthesised protein plus/minus one standard deviation. (f)
Microarrays of T. brucei
proteins were incubated with the labelled, synthetic RNA sequences
shown. The
white circles highlight positive signals. On the lower array, also a
second
protein exhibited interaction. In (g),
the interacting protein from the lower microarray was analysed in more
detail
in comparison to a derivative with one point mutation (Mut) and
another,
unrelated protein. The upper level shows the protein amounts, detected
by
antibody binding to the N-terminus. The lower panel shows binding of
the
synthetic RNA. Subsequent studies demonstrated a more than 3,000-fold
difference in affinity to the specific RNA between wildtype and mutated
protein. (h) Binding to 74 human
transcription factors of a labelled synthetic DNA sequence representing
the
TERT promoter (left). Mutation of one base pair in the binding sequence
led to
a very different binding pattern (right).
..
Third
panel: Detection of proteins generated in situ
from individual samples. (i) Quality assessment of
on-chip PCR by oligonucleotide
hybridisation; two oligonucleotides were used, labelled red or green,
respectively, each binding to one of the DNA-strands. Typical results
are shown
with an oligonucleotide specific to only one PCR-product present in
quadruplicate (left) and a simultaneous hybridisation with
oligonucleotides to
all PCR-products (right). (j) Fusion
of the images obtained after an incubation with fluorescently labelled
antibodies against N- (red signal) and C-terminal tags (green signal)
of the
expressed seven tumour marker proteins. Spots 8 and 9 were negative
controls
without DNA-template. (k, l) Protein
detection with labelled
antibodies that target proteins CDK2 and TP53, respectively. (m) Results obtained on arrays produced
from tissue samples of individual patients. Normal = healthy pancreas;
CP =
chronic pancreatitis; PDAC = pancreatic ductal adenocarcinoma;
MiaPaca-2 = PDAC
cell line. All proteins were identified with a tag-specific antibody
(left).
Binding patterns obtained with two different, isoform-specific
antibodies. One
isoform of the RUNX1 protein was present in all samples (right); the
other one
was found in diseased material only (middle). (n)
Ninety-six DARPin binders were expressed in situ, each
in three copies. Tag-specific antibodies identified
all binders (left). The other two panels (middle, right) show binding
patterns
obtained upon incubation with protein AKT3. The white frames indicate
the 16
binders that were expected to interact with AKT3. Different washing
stringency
produced distinct variations in the binding patterns.
|
|
Syafrizayanti et
al. (2017) Sci. Rep.
7, 39756. |
.
Gene expression regulatory networks
in Trypanosoma brucei:
insights into the role of the mRNA binding proteome
Control
of gene expression at the
post-transcriptional level is essential in all organisms, and
RNA-binding
proteins play critical roles from mRNA synthesis to decay. To fully
understand
this process, it is necessary to identify the complete set of
RNA-binding
proteins and the functional consequences of the protein-mRNA
interactions.
Here, we provide an overview of the proteins that bind to mRNAs and
their
functions in the pathogenic bloodstream form of Trypanosoma
brucei. We describe the production of a small
collection of open-reading frames encoding proteins potentially
involved in
mRNA metabolism. With this ORFeome collection, we used tethering to
screen for
proteins that play a role in post-transcriptional control. A yeast
two-hybrid
screen showed that several of the discovered repressors interact with
components of the CAF1/NOT1 deadenylation complex. To identify the
RNA-binding
proteins, we obtained the mRNA-bound proteome. We identified 155
high-confidence candidates, including many not previously annotated as
RNA-binding proteins. Twenty seven of these proteins affected reporter
expression in the tethering screen. Our study provides novel insights
into the
potential trypanosome mRNPs composition, architecture and function
Lueong et
al. (2016) Mol.
Microbiol. 100,
457-471.
Erben et
al. (2014) PLOS
Pathogens 10,
e1004178. |
|
Nine
microarrays containing proteins expressed from
5 pg each of some 14,000 PCR-products.
The microarray-bound proteins were
labelled with luminescent dye for quality control purposes
|
Combinatorial
peptide synthesis with laser-based
transfer of monomers

Laser writing is used to
structure surfaces in many different ways in materials and life
sciences.
However, combinatorial patterning applications are still limited. In a
collaborative project coordinated by colleagues at KIT, a method was
developed for cost-efficient combinatorial synthesis of
very-high-density peptide
arrays with natural and synthetic monomers. A laser automatically
transfers
nanometre-thin solid material spots from different donor slides to an
acceptor.
Each donor bears a thin polymer film, embedding one type of monomer.
Coupling
occurs in a separate heating step, where the matrix becomes viscous and
building blocks diffuse and couple to the acceptor surface.
Furthermore, two
material layers of activation reagents and amino acids can be deposited
consecutively.
Subsequent heat-induced mixing facilitates an in situ
activation and
coupling of the monomers. This allows to incorporate building blocks
with click
chemistry compatibility or a large variety of commercially available
non-activated, for example, post-translationally modified
building blocks into the array’s peptides with >17,000 spots per
square
centimetre.
Loeffler et
al. (2016) Nature Comm. 7,
11844.
|
|