Functional
Genome Analysis (B070)
Deutsches
Krebsforschungszentrum,
Im Neuenheimer Feld 580
D-69120
Heidelberg,
Germany. |
 |
..
.
..
Transcript
Studies
..
...
| For the understanding of the complex
regulative mechanisms and the investigation of the cellular
control management, a simultaneous analysis of the expression of all
genes of an organism under various conditions and over time is
indispensable. Our
emphasis is on studying human cancer material and analyses particularly at
the level of
microRNA, with other RNA types also being worked at,
although to a lesser extent.
Information is gathered for enabling
early
diagnosis and accurate prognosis, the identification of potentially
interesting
avenues for therapy as well as the evaluation of the success of disease
treatment. For this, the measurements of transcriptional variations are
combined with the analysis of epigenetic modulations of the
genomic DNA
and actual protein expression. For
particular microRNA molecules, their mode of action is studied in
detail, elucidating the mechanisms by which their activity is
transformed into function. |
Blood-based
diagnosis and risk stratification of patients with intraductal
papillary mucinous neoplasm (IPMN) to decide on surgical intervention
Intraductal
papillary mucinous neoplasm (IPMN) is a
precursor of PDAC. Patients with low-grade dysplasia have a relatively
good
prognosis and are kept under surveillance to monitor disease
development,
whereas high-grade dysplasia and IPMN invasive carcinoma require tumour
resection. Diagnostic distinction of the two groups is difficult,
however.
We
aimed to identify variations in protein concentration in peripheral
blood for
accurate discrimination. Sera from IPMN patients and healthy donors were
analysed on microarrays made of antibodies for studying protein level
variations. For microRNA (miRNA) biomarkers, a PCR-based screen was
performed
and
biomarker candidates confirmed by quantitative PCR.
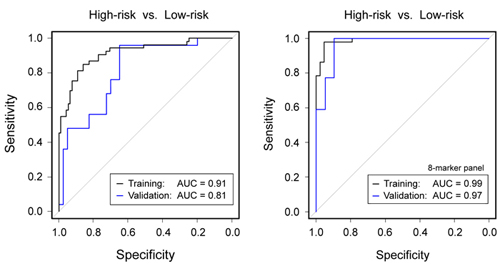
A support vector
machine
(SVM) algorithm defined classifiers, which were validated on a separate
sample
set. A panel of five proteins and three miRNAs could distinguish high-
and
low-risk IPMN with an accuracy of 97%. This is substantially better
than the
accuracy obtained in the same patient cohort by using the guideline
criteria
for decision-making on performing surgery or not. The precise
blood-based
diagnosis and risk stratification will improve patient management and
thus the
prognosis of IPMN patients. In addition to the main finding, highly
accurate
discrimination was also achieved between other patient subgroups.
Zhang
et
al.
(2023) Clin. Cancer
Res. 29, 1535-1545. 
|
|
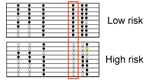
Figure legend: (Left) Diagnostic
performance of
clinical parameters to discriminate high-risk from low-risk IPMN
according to
current guidelines. (Right) Much
better results were obtained by a combined panel of 5 protein and 3
miRNA
biomarkers. The results are presented as ROC curves and corresponding
AUC
values as determined in the training and validation cohorts,
respectively. |
.
..
 |
|
|
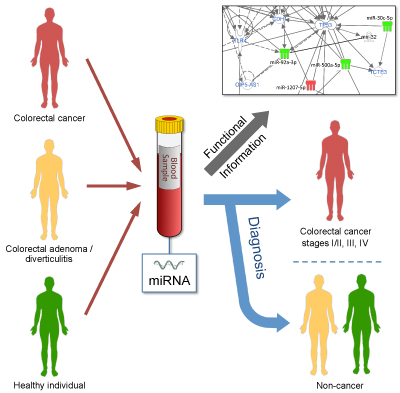
MicroRNAs in
blood act as biomarkers of colorectal cancer and indicate potential
therapeutic targets
Association studies have linked alterations of
blood-derived microRNAs
(miRNAs) with colorectal cancer (CRC). We performed a microarray-based
comparison of the profiles of 2,549 miRNAs in 80 blood samples from
healthy
donors and patients with colorectal adenomas, colorectal diverticulitis
and CRC
at different stages. Confirmation by quantitative real-time PCR
(RT-PCR) was
complemented by validation of identified molecules in another 36 blood
samples.
No variations in miRNA levels were observed in samples
from patients with
colorectal adenomas and diverticulitis or from healthy donors. However,
there
were 179 CRC-associated miRNAs of differential abundance compared to
healthy
controls. Only three – miR-1225-5p, miR-1207-5p and miR-4459 –
exhibited
increased levels at all CRC stages. Most deregulated miRNAs (128/179,
71%)
specifically predicted metastatic CRC. Pathway analysis found several cancer-related pathways to which
the miRNAs
contribute in various ways. In conclusion, miRNA levels in blood vary
throughout CRC progression and affect cellular functions relevant to
haematogenous CRC progression and dissemination.
rr
Stang et al. (2021) Mol. Oncol. 15, 2480-2490.
|
|
..
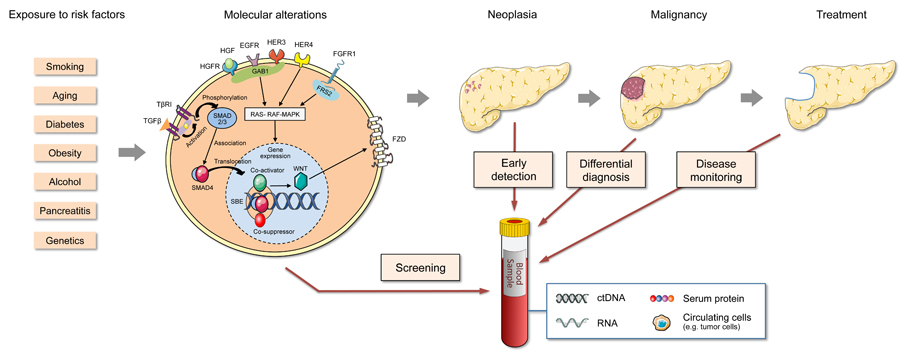
Blood biomarkers
for differential diagnosis and early detection of pancreatic cancerr
Pancreatic cancer is currently
the most lethal tumour entity and case numbers are rising. It
will soon
be the second most frequent cause of cancer-related death in the
Western world.
Mortality is close to incidence and patient survival after diagnosis
stands at
about five months.
..
Blood-based diagnostics could be a crucial factor for
improving this dismal situation and is at a stage that could make this
possible. We reviewed in much detail the current state of affairs with
its
problems and promises, looking at various molecule types including
microRNAs.
Reported results were evaluated in the overall context. Also, we
proposed steps
toward clinical utility that should advance the development toward
clinical
application by improving biomarker quality but also by defining
distinct clinical
objectives and the respective diagnostic accuracies required to achieve
them.
Many of the discussed points and conclusions are highly relevant to
other solid
tumours, too.
Al-Shaheri et al. (2021) Cancer Treat. Rev. 96, 102193.
|
 |
|
|
,
Figure legend. Aspects of PDAC tumour
development and possible application areas of liquid biopsy
diagnostics. Many
processes during tumorigenesis result in molecular changes that may be
detectable in peripheral blood. After transformation, factors released
for
niche formation or as part of the molecular communication between cells
of the
tumour microenvironment could circulate in the blood and permit
detection at
early stages. Furthermore, such biomarkers could provide valuable
information
for a differential diagnosis, discriminating malign from benign
diseases, for
example, or allowing therapy monitoring.
|
|
|
|