Functional
Genome Analysis (B070)
Deutsches
Krebsforschungszentrum,
Im Neuenheimer Feld 580
D-69120
Heidelberg,
Germany. |
 |
.
..
DNA / RNA Technologies
..
..
|
|
Genome-wide
CRISPR-Cas9 knockout screen reveals genes with critical role in
specifically metastatic PDAC cells
Early and aggressive
metastasis is a crucial factor of the dismal prognosis of pancreatic
ductaladenocarcinoma (PDAC). Toward
identifying processes that are essential to PDAC metastasis, we
performed a
genome-wide gene knockout screen with a CRISPR-Cas9 library of 259,900 single guide RNA constructs on two sub-clones of a PDAC cell line, which
exhibit very low or very high metastatic potential, respectively.
Data
comparison identified several genes and pathways that were relevant for
metastasising cells. One knockout had a particularly strong effect and
was analysed with respect to functional aspects. It was critical for
the
difference in metastatic potential. Also, a buffering interaction was found between this gene and a
master regulator
in cancer.
In vivo
experiments, namely
lung and liver colonisation assays after intravenous and intracardiac
cell
injections into the bloodstream as well as metastatic behaviour after
resection
of subcutaneously grown primary tumours, confirmed the very
strong
effect on metastasis.
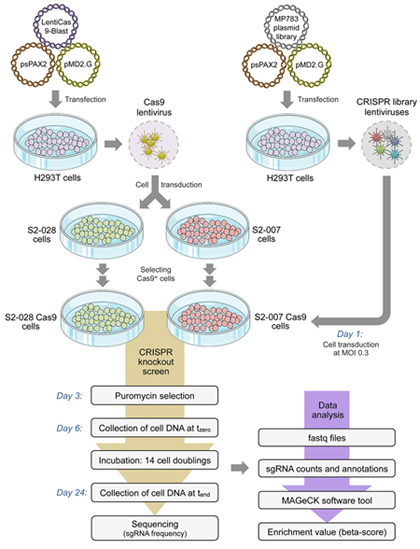
Figure legend:
Schematic
outline of the genome-wide knockout experiment.
|
|
...
Process for an
efficient lentiviral cell transduction
The combination of lentiviruses with techniques such as
CRISPR-Cas9 has resulted in efficient and precise processes for
targeted genome
modification. An often-limiting aspect, however, is the efficiency of
cell
transduction. Low efficiencies with particular cell types and/or the
high
complexity of lentiviral libraries can cause insufficient
representation.
We
have developed a protocol that yielded substantial increases in
transduction
efficiency in various cell lines in comparison to several other
procedures. By
applying and combining relatively simple procedural
modifications – (i) mixing lipofectamines
3000 and LTX for transfecting HEK-293T cells,
(ii) virus
concentration in spin-columns and (iii) gently pelleting cells prior to
transfection –
substantial improvements
in transduction yields could be achieved.
Pirona et al. (2020) Biol. Method Protoc., bpaa005. 
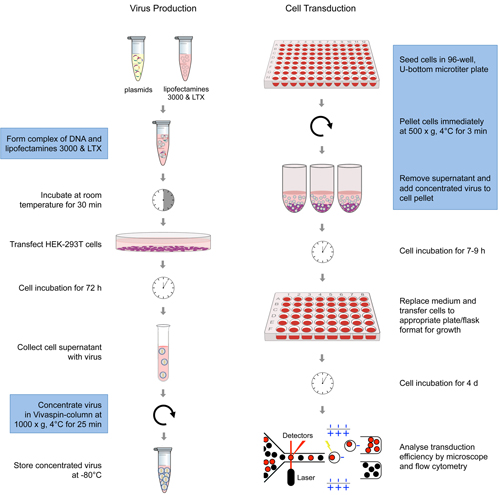
Figure legend: Schematic overview of the various steps of the
optimised transduction process. The
steps in blue-labelled frames are different to standard protocols and
jointly
increase transduction efficiency substantially
|
|
|
|
...
CRISPR
RNA-guided FokI nucleases
repair a disease-causing variant in the PAH gene in a phenylketonuria model
In a proof-of-concept
study, we demonstrated the potential of an improved CRISPR/Cas9 system – the FokI-dCas9
system – for
precision medicine, in particular for targeting phenylketonuria
(PKU) and other
monogenic metabolic diseases. The FokI-dCas9
system can greatly
improve the specificity of genome editing. In contrast to the
standard system, it requires dimerization of the FokI-dCas9-sgRNA
complex,
meaning that monomeric FokI-dCas9-sgRNA is unable to cut the
DNA strand,
thus reducing substantially the chances of contaminating off-target
effects.
..
PKU
is the most common inherited disease in amino acid metabolism. It leads
to
severe neurological and neuropsychological symptoms if untreated or
late
diagnosed. Correction of the disease-causing variants in the
phenylalanine
hydroxylase (PAH) gene could rescue residual activity and
restore normal
function. The CRISPR/Cas9 system is a recently developed genome editing
technique. We applied a modification, which employs the fusion of
inactive Cas9
(dCas9) and the FokI endonuclease (FokI-dCas9) to
correct the
most common variant (allele frequency 21.4%) in the PAH gene -
c.1222C>T (p.Arg408Trp) - as an approach toward curing PKU. Co-expression
of a single guide RNA plasmid, a FokI-dCas9-zsGreen1 plasmid,
and the
presence of a single-stranded oligodeoxynucleotide in PAH_c.1222C>T
COS-7 cells – an in vitro model of PKU – corrected the PAH
variant
and restored PAH activity.
Pan et al. (2016) Sci. Rep. 6, 35794. |
|
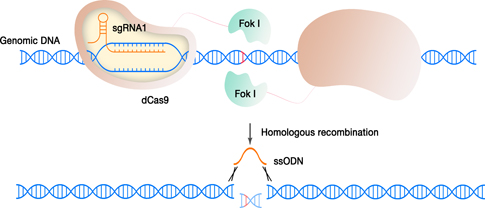
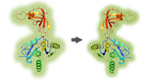
Scheme of the process: one dimer of the FokI-dCas9
complex binds to two “half-sites” on the genome with a certain spacer
length
and generate double-strand breaks in the DNA. The double-strand breaks
are then
repaired by homology directed repair, introducing the non-mutated
sequence provided as an oligonucleotide.
|
|
...
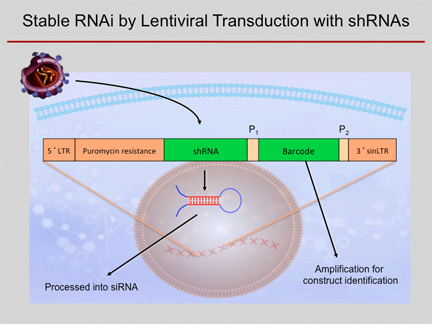
Scheme of shRNA
knockdown experiments. An shRNA-construct is brought into a cell by
means of a lentivirus system. The construct integrates into the genome
and the respective shRNA is constitutively expressed at high levels,
acting as an inhibitory RNA intracellularly. Using a common primer pair
(P1, P2), a barcode sequence that is unique for each shRNA construct can be amplified from
each cell. Therefore,
also complex mixtures of cells transduced with different constructs can
be studied simultaneously.
|
|
Functional
screens by means of lentiviral shRNA libraries
RNA
interference (RNAi) has become a popular and important tool for the
analysis of gene
function, although being partly superseeded by CRISPR-Cas already.
Loss-of-function studies have greatly facilitated functional analyses
of the
human transcriptome.
...
Limitations of early siRNA experiments, most
importantly the transient inhibition of gene expression as well
as the
inefficient transfection into non-dividing cells, were generally
overcome by short hairpin RNA (shRNA)
expression
vectors, which
stably integrate into a target cell's genome via
retro-
or lentiviral gene transfer. Intracellular processing of shRNAs results
in
short duplex RNAs with siRNA-like properties. Viral integration ensures
not
only a broad range of infectable target cell types but also the stable
expression of specific shRNAs, resulting in the permanent reduction of
the
targeted gene product. Complex shRNA expression libraries
allow the targeted knockdown of
thousands
of different genes in a single experiment.
...
Using
such lentiviral vector
shRNA libraries and
initially barcode arrays and meanwhile next-generation sequencing analysis for decoding of the pooled RNAi screens, we are able to quantify the
abundance of
individual shRNAs and thus determine in a complex pool the number of
cells infected with an individual shRNA construct.
...
We used the
technique to
predict anti-proliferative effects of individual shRNAs from pooled
negative
selection screens. By such screens, we identified synthetic-lethal
activities toward combination therapies, defined genes which are
required
for a stem-cell
like phenotype and found tumour
suppressor genes by in vivo
studies. Further
studies are under way, both for the elucidation of basic regulative
processes associated to cancer and for the identification of pathways
that are affected by particular drugs or compounds. In particular, we
use the technique for obtaining more detailed information
on the functional effects of particularly potentially druggable gene
products.
|
|
Wolf et al. (2014) Oncogene 33, 4273-4278. |
|
|
Fredebohm et al. (2013) J. Cell Sci. 126, 3380-3389. |
|
|
|
Böttcher et al. (2014) BMC Genomics 15,
158. |
|
|
Böttcher et al. (2010) BMC Genomics 11, 7. |
 |
|
|
Wolf et al. (2013) Breast Cancer Res. 15, R109. |
|
|
Böttcher & Hoheisel (2010) Curr. Genom. 11, 162-167. |
|
|
|
|
|